Haynes Chad, Oldfield Christopher J, Ji Fei, Klitgord Niels, Cusick Michael E, Radivojac Predrag, Uversky Vladimir N, Vidal Marc, Iakoucheva Lilia M
Laboratory of Statistical Genetics, The Rockefeller University, New York, New York, USA.
PLoS Comput Biol. 2006 Aug 4;2(8):e100. doi: 10.1371/journal.pcbi.0020100. Epub 2006 Jun 23.
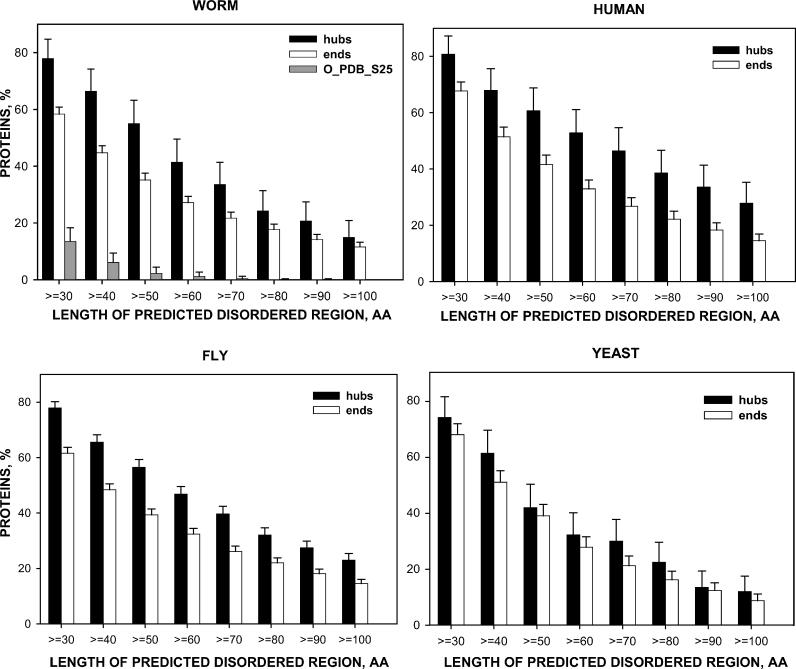
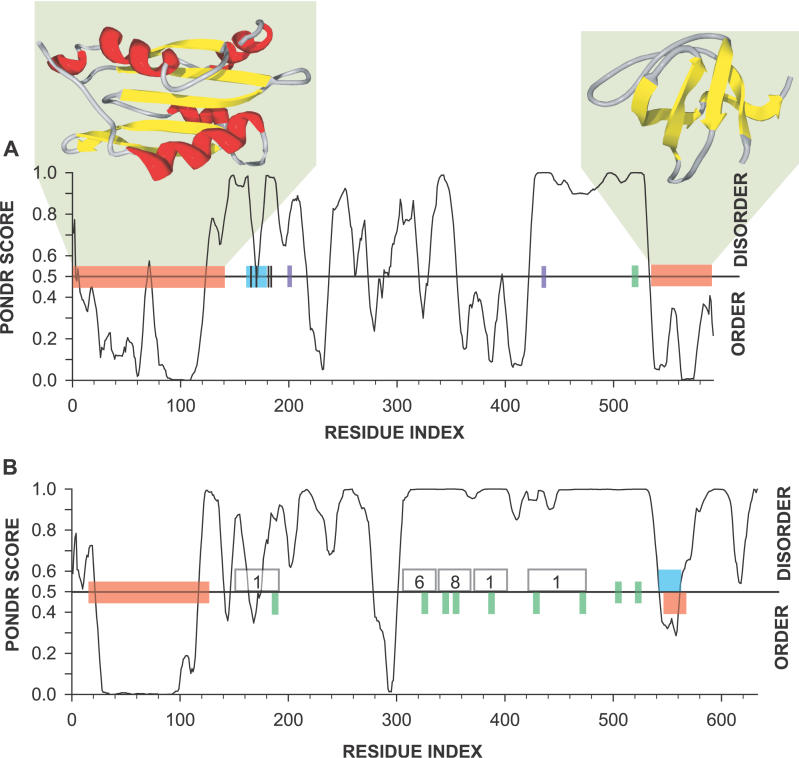
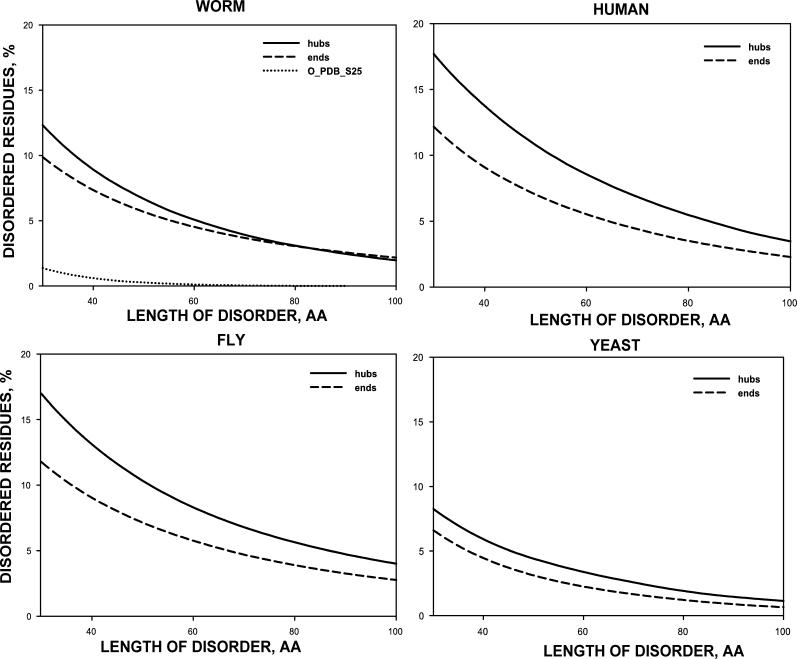
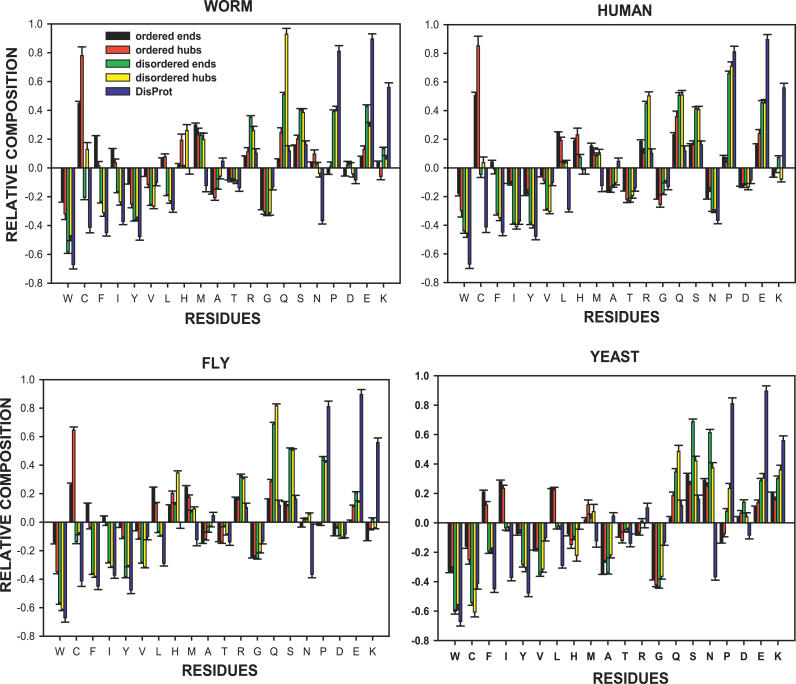
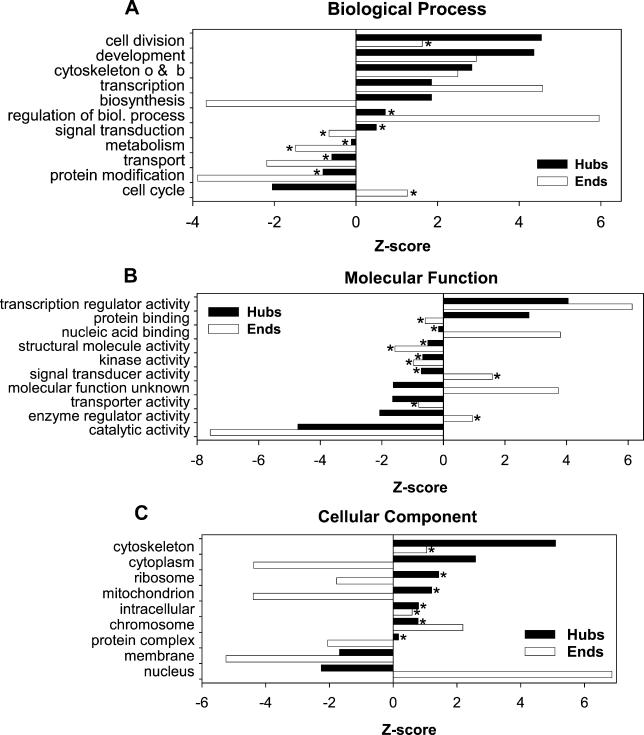
Recent proteome-wide screening approaches have provided a wealth of information about interacting proteins in various organisms. To test for a potential association between protein connectivity and the amount of predicted structural disorder, the disorder propensities of proteins with various numbers of interacting partners from four eukaryotic organisms (Caenorhabditis elegans, Saccharomyces cerevisiae, Drosophila melanogaster, and Homo sapiens) were investigated. The results of PONDR VL-XT disorder analysis show that for all four studied organisms, hub proteins, defined here as those that interact with > or = 10 partners, are significantly more disordered than end proteins, defined here as those that interact with just one partner. The proportion of predicted disordered residues, the average disorder score, and the number of predicted disordered regions of various lengths were higher overall in hubs than in ends. A binary classification of hubs and ends into ordered and disordered subclasses using the consensus prediction method showed a significant enrichment of wholly disordered proteins and a significant depletion of wholly ordered proteins in hubs relative to ends in worm, fly, and human. The functional annotation of yeast hubs and ends using GO categories and the correlation of these annotations with disorder predictions demonstrate that proteins with regulation, transcription, and development annotations are enriched in disorder, whereas proteins with catalytic activity, transport, and membrane localization annotations are depleted in disorder. The results of this study demonstrate that intrinsic structural disorder is a distinctive and common characteristic of eukaryotic hub proteins, and that disorder may serve as a determinant of protein interactivity.
近期的全蛋白质组筛选方法已经提供了大量关于各种生物体中相互作用蛋白质的信息。为了测试蛋白质连接性与预测的结构无序程度之间的潜在关联,研究了来自四种真核生物(秀丽隐杆线虫、酿酒酵母、黑腹果蝇和智人)的具有不同数量相互作用伙伴的蛋白质的无序倾向。PONDR VL-XT无序分析结果表明,对于所有四种研究的生物体,这里定义为与≥10个伙伴相互作用的中枢蛋白,比这里定义为仅与一个伙伴相互作用的末端蛋白明显更无序。中枢蛋白中预测的无序残基比例、平均无序得分以及不同长度的预测无序区域数量总体上高于末端蛋白。使用一致性预测方法将中枢蛋白和末端蛋白二元分类为有序和无序亚类,结果显示,相对于蠕虫、果蝇和人类中的末端蛋白,中枢蛋白中完全无序的蛋白质显著富集,完全有序的蛋白质显著减少。使用基因本体论(GO)类别对酵母中枢蛋白和末端蛋白进行功能注释,以及这些注释与无序预测的相关性表明,具有调控、转录和发育注释的蛋白质在无序方面富集,而具有催化活性、转运和膜定位注释的蛋白质在无序方面减少。这项研究的结果表明,内在结构无序是真核生物中枢蛋白的一个独特且常见的特征,并且无序可能是蛋白质相互作用的一个决定因素。