Jia D, O'Brien C A, Stewart S A, Manolagas S C, Weinstein R S
Department of Internal Medicine, and the Central Arkansas Veterans Healthcare System, University of Arkansas for Medical Sciences, 4301 West Markham Street, Slot 587, Little Rock, Arkansas 72205, USA.
Endocrinology. 2006 Dec;147(12):5592-9. doi: 10.1210/en.2006-0459. Epub 2006 Aug 24.
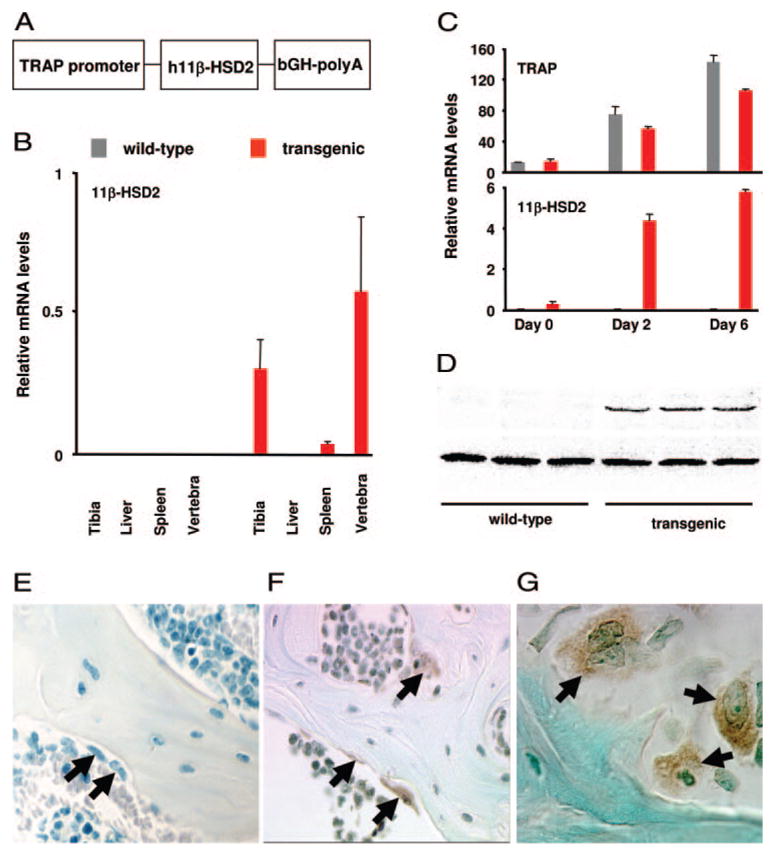
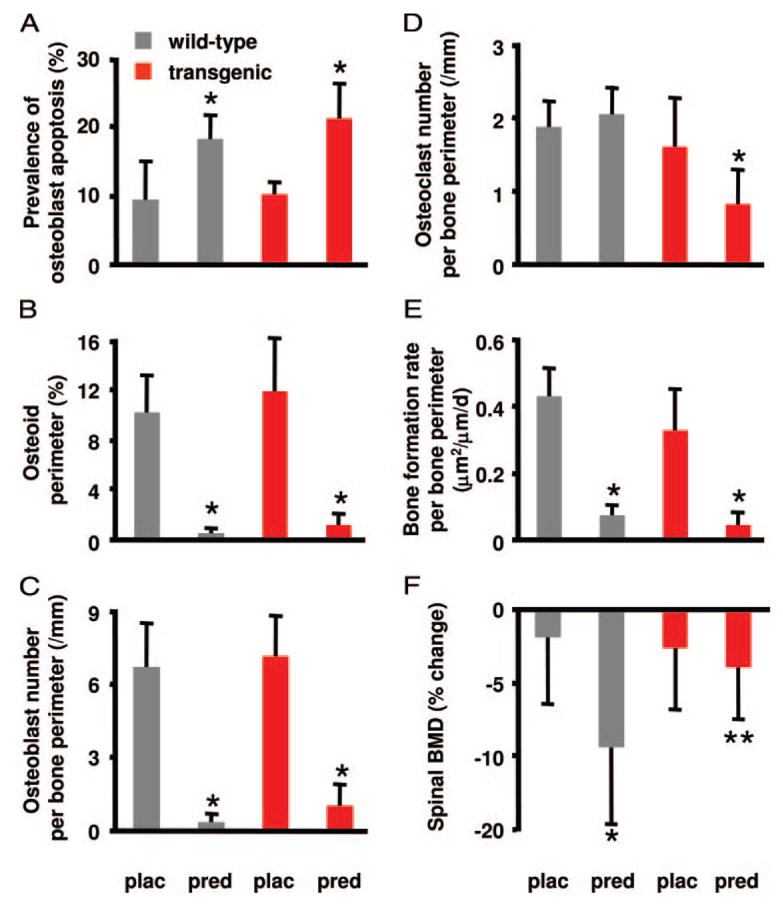
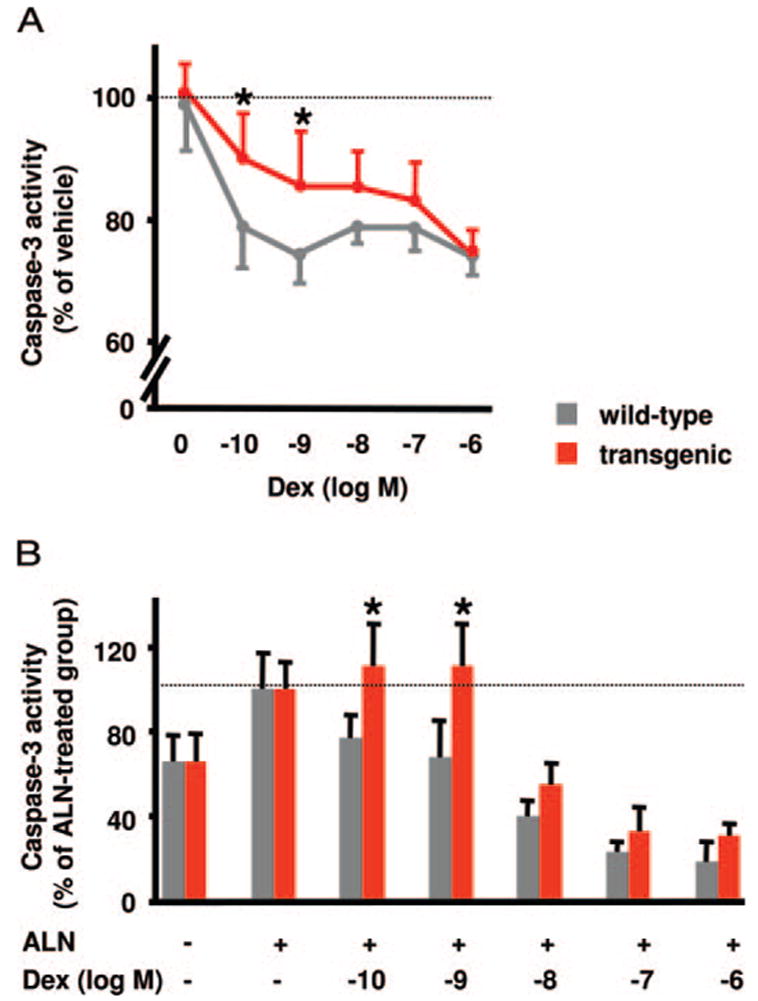
Glucocorticoid administration to mice results in a rapid loss of bone mineral density due to an imbalance in osteoblast and osteoclast numbers. Whereas excess glucocorticoids reduce both osteoblast and osteoclast precursors, cancellous osteoclast number surprisingly does not decrease as does osteoblast number, presumably due to the ability of glucocorticoids to promote osteoclast life span. Whether glucocorticoids act directly on osteoclasts in vivo to promote their life span and whether this contributes to the rapid loss of bone with glucocorticoid excess remains unknown. To determine the direct effects of glucocorticoids on osteoclasts in vivo, we expressed 11beta-hydroxysteroid dehydrogenase type 2, an enzyme that inactivates glucocorticoids, specifically in the osteoclasts of transgenic mice using the tartrate-resistant acid phosphatase promoter. Bone mass, geometry, and histomorphometry were similar in untreated wild-type and transgenic animals. Glucocorticoid administration for 7 d caused equivalent increases in cancellous osteoblast apoptosis, and equivalent decreases in osteoblasts, osteoid, and bone formation, in wild-type and transgenic mice. In contrast, glucocorticoids stimulated expression of the mRNA for calcitonin receptor, an osteoclast product, in wild-type but not transgenic mice. Consistent with the previous finding that glucocorticoids decrease osteoclast precursors and prolong osteoclast life span, glucocorticoids decreased cancellous osteoclast number in the transgenic mice but not wild-type mice. In accord with this decrease in osteoclast number, the loss of bone density observed in wild-type mice was strikingly prevented in transgenic mice. These results demonstrate for the first time that the early, rapid loss of bone caused by glucocorticoid excess results from direct actions on osteoclasts.
给小鼠施用糖皮质激素会导致骨矿物质密度迅速下降,这是由于成骨细胞和破骨细胞数量失衡所致。虽然过量的糖皮质激素会减少成骨细胞和破骨细胞前体,但松质骨破骨细胞数量出人意料地不像成骨细胞数量那样减少,推测这是因为糖皮质激素具有延长破骨细胞寿命的能力。糖皮质激素在体内是否直接作用于破骨细胞以延长其寿命,以及这是否导致糖皮质激素过量时骨的快速流失,目前尚不清楚。为了确定糖皮质激素在体内对破骨细胞的直接作用,我们利用抗酒石酸酸性磷酸酶启动子,在转基因小鼠的破骨细胞中特异性表达2型11β-羟基类固醇脱氢酶,该酶可使糖皮质激素失活。未处理的野生型和转基因动物的骨量、几何形状和组织形态计量学相似。野生型和转基因小鼠连续7天施用糖皮质激素后,松质骨成骨细胞凋亡均有同等程度增加,成骨细胞、类骨质和骨形成均有同等程度减少。相反,糖皮质激素刺激野生型而非转基因小鼠中降钙素受体(一种破骨细胞产物)的mRNA表达。与之前发现的糖皮质激素减少破骨细胞前体并延长破骨细胞寿命一致,糖皮质激素使转基因小鼠而非野生型小鼠的松质骨破骨细胞数量减少。与破骨细胞数量的减少一致,转基因小鼠显著预防了野生型小鼠中观察到的骨密度损失。这些结果首次证明,糖皮质激素过量导致的早期、快速骨流失是由其对破骨细胞的直接作用引起的。