Adachi Jun, Kumar Chanchal, Zhang Yanling, Olsen Jesper V, Mann Matthias
Department of Proteomics and Signal Transduction, Max-Planck Institute for Biochemistry, Am Klopferspitz, D-82152 Martinsried, Germany.
Genome Biol. 2006;7(9):R80. doi: 10.1186/gb-2006-7-9-R80.
Urine is a desirable material for the diagnosis and classification of diseases because of the convenience of its collection in large amounts; however, all of the urinary proteome catalogs currently being generated have limitations in their depth and confidence of identification. Our laboratory has developed methods for the in-depth characterization of body fluids; these involve a linear ion trap-Fourier transform (LTQ-FT) and a linear ion trap-orbitrap (LTQ-Orbitrap) mass spectrometer. Here we applied these methods to the analysis of the human urinary proteome.
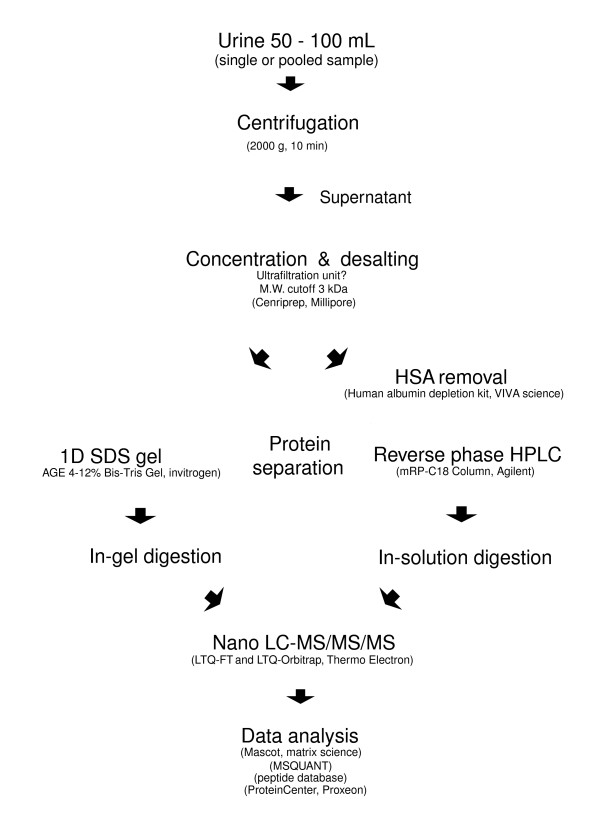
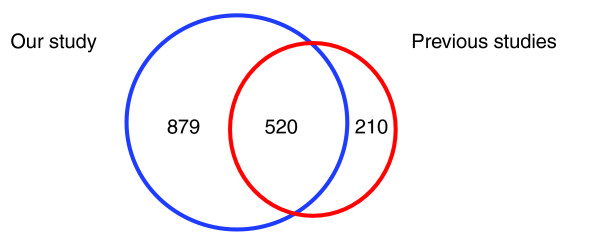
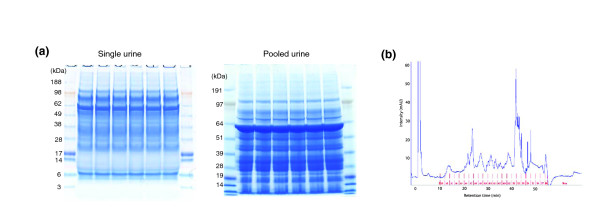
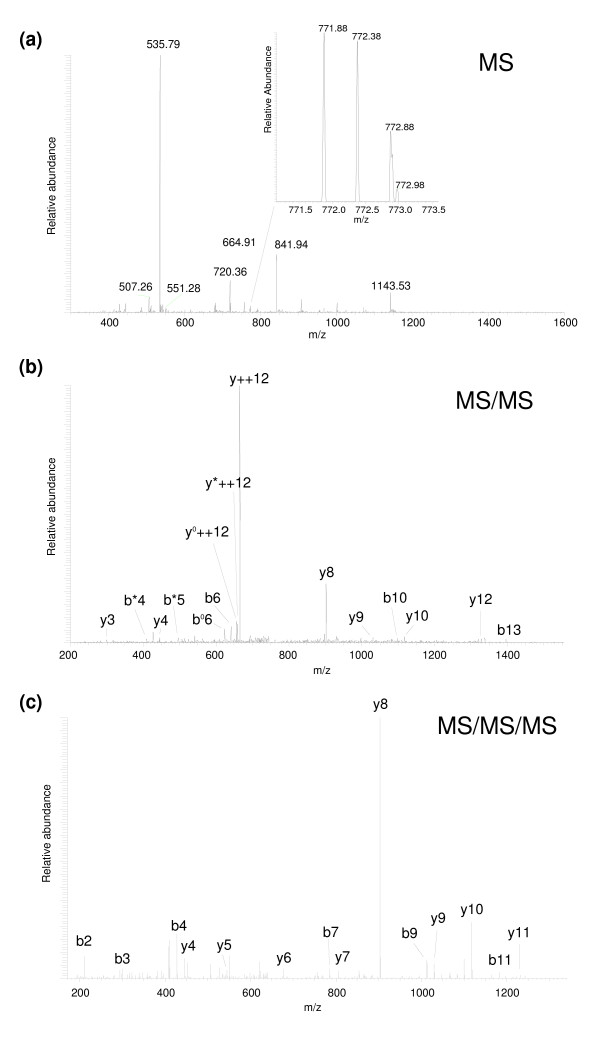
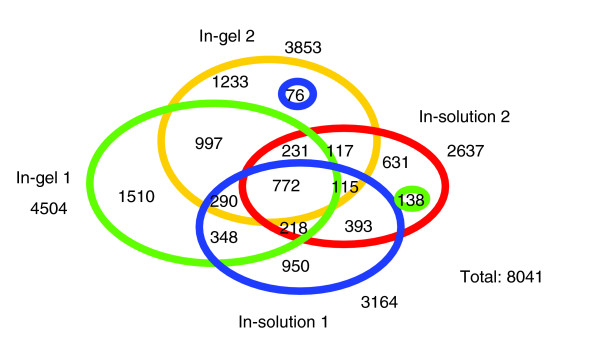
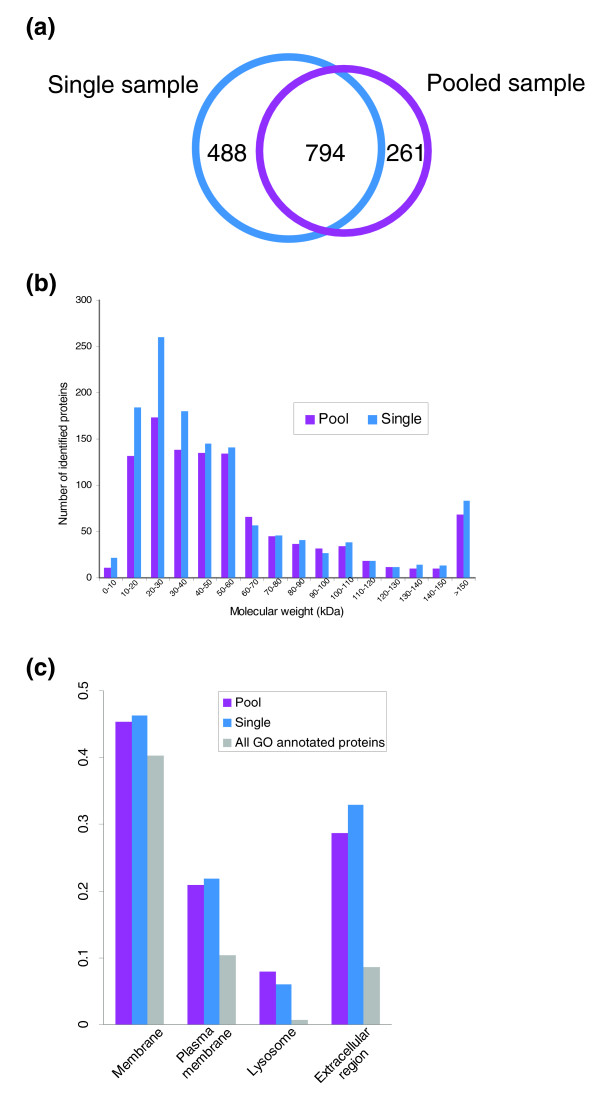
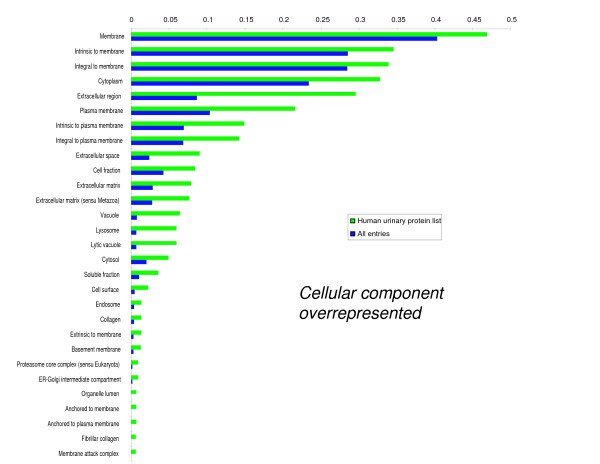
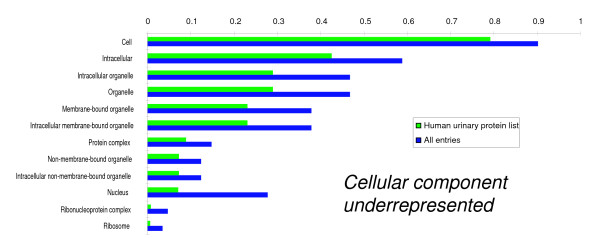
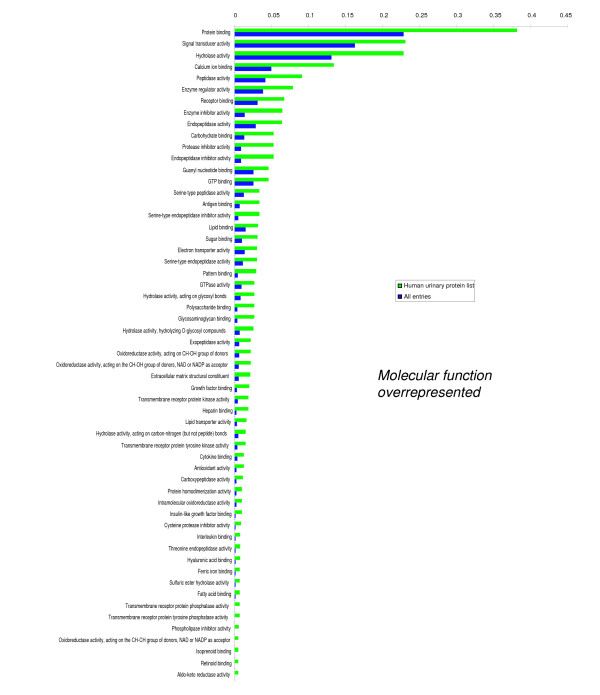
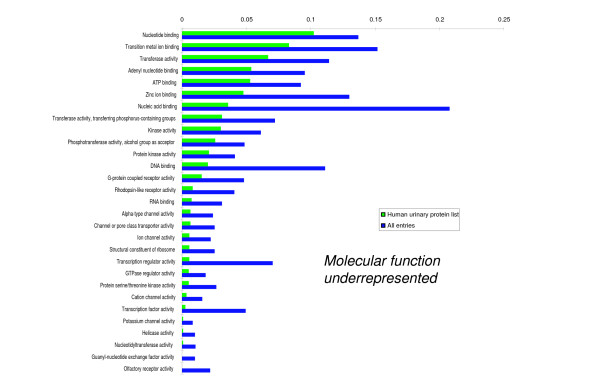
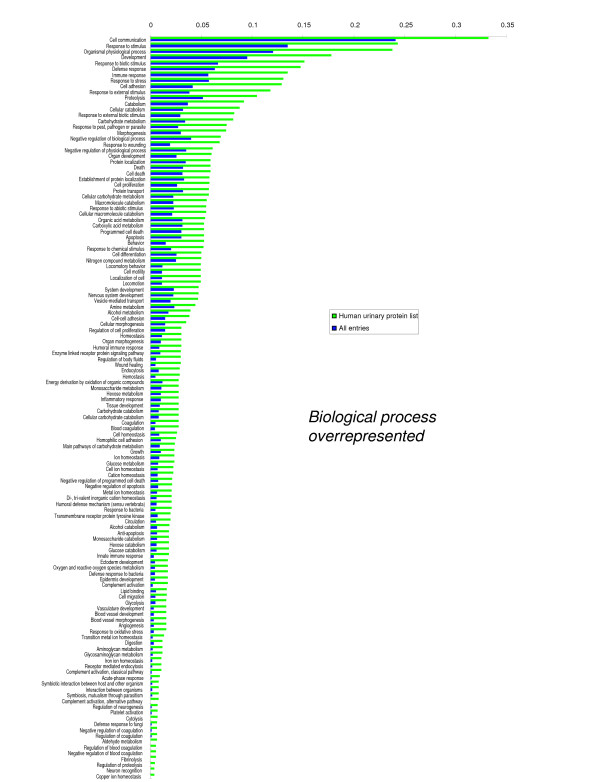
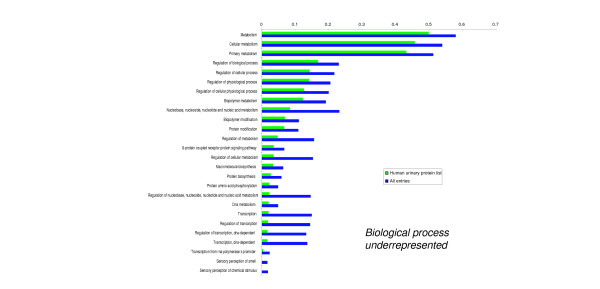
We employed one-dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis and reverse phase high-performance liquid chromatography for protein separation and fractionation. Fractionated proteins were digested in-gel or in-solution, and digests were analyzed with the LTQ-FT and LTQ-Orbitrap at parts per million accuracy and with two consecutive stages of mass spectrometric fragmentation. We identified 1543 proteins in urine obtained from ten healthy donors, while essentially eliminating false-positive identifications. Surprisingly, nearly half of the annotated proteins were membrane proteins according to Gene Ontology (GO) analysis. Furthermore, extracellular, lysosomal, and plasma membrane proteins were enriched in the urine compared with all GO entries. Plasma membrane proteins are probably present in urine by secretion in exosomes.
Our analysis provides a high-confidence set of proteins present in human urinary proteome and provides a useful reference for comparing datasets obtained using different methodologies. The urinary proteome is unexpectedly complex and may prove useful in biomarker discovery in the future.
尿液因其易于大量收集,是疾病诊断和分类的理想材料;然而,目前生成的所有尿液蛋白质组目录在鉴定深度和可信度方面都存在局限性。我们实验室已开发出对体液进行深度表征的方法;这些方法涉及线性离子阱-傅里叶变换(LTQ-FT)和线性离子阱-轨道阱(LTQ-Orbitrap)质谱仪。在此,我们将这些方法应用于人类尿液蛋白质组的分析。
我们采用一维十二烷基硫酸钠聚丙烯酰胺凝胶电泳和反相高效液相色谱进行蛋白质分离和分级。分级后的蛋白质在凝胶内或溶液中进行消化,消化产物用LTQ-FT和LTQ-Orbitrap以百万分之一的精度并通过两个连续阶段的质谱碎裂进行分析。我们从十名健康供体获取的尿液中鉴定出1543种蛋白质,同时基本消除了假阳性鉴定。令人惊讶的是,根据基因本体论(GO)分析,近一半的注释蛋白质是膜蛋白。此外,与所有GO条目相比,尿液中细胞外、溶酶体和质膜蛋白有所富集。质膜蛋白可能通过外泌体分泌存在于尿液中。
我们的分析提供了一组存在于人类尿液蛋白质组中的高可信度蛋白质,并为比较使用不同方法获得的数据集提供了有用的参考。尿液蛋白质组出人意料地复杂,未来可能在生物标志物发现中发挥作用。