Titulaer Mark K, Siccama Ivar, Dekker Lennard J, van Rijswijk Angelique L C T, Heeren Ron M A, Sillevis Smitt Peter A, Luider Theo M
Department of Neurology, Erasmus MC, Rotterdam, The Netherlands.
BMC Bioinformatics. 2006 Sep 5;7:403. doi: 10.1186/1471-2105-7-403.
Statistical comparison of peptide profiles in biomarker discovery requires fast, user-friendly software for high throughput data analysis. Important features are flexibility in changing input variables and statistical analysis of peptides that are differentially expressed between patient and control groups. In addition, integration the mass spectrometry data with the results of other experiments, such as microarray analysis, and information from other databases requires a central storage of the profile matrix, where protein id's can be added to peptide masses of interest.
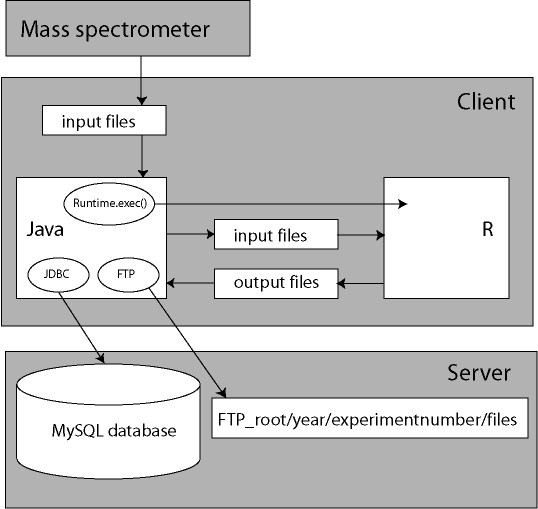
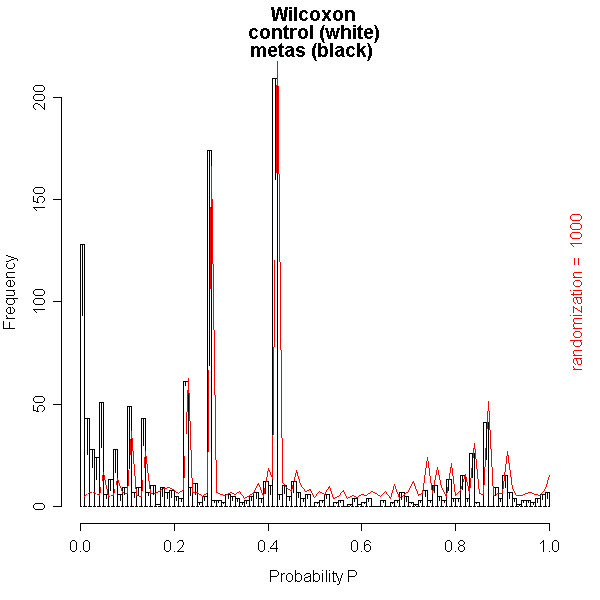
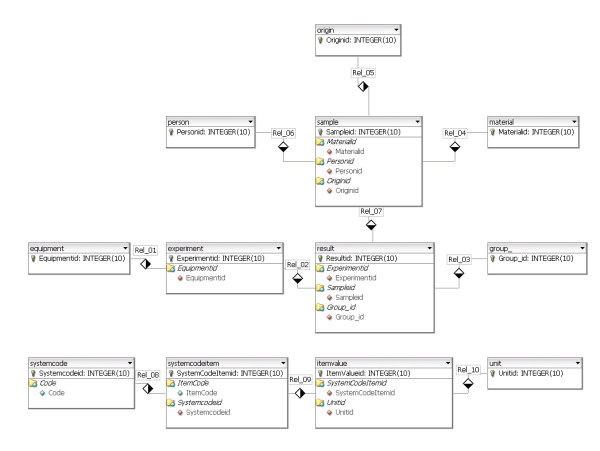
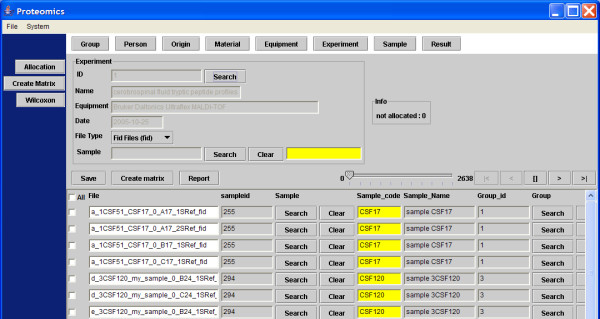
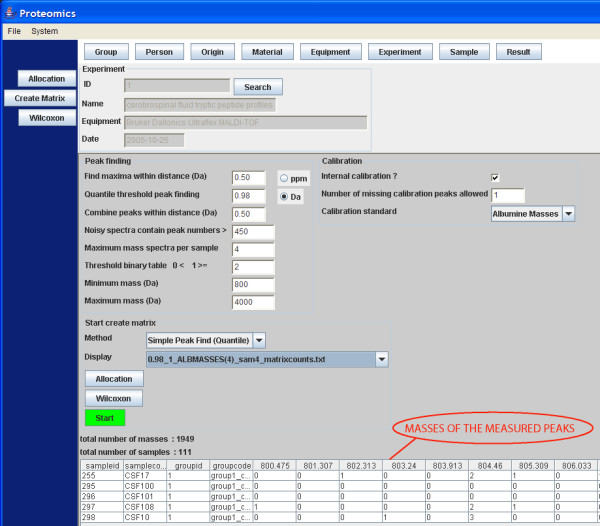
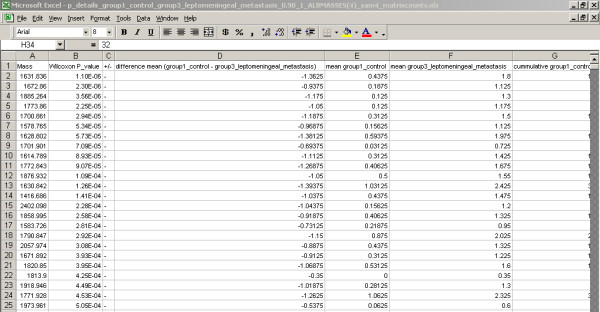
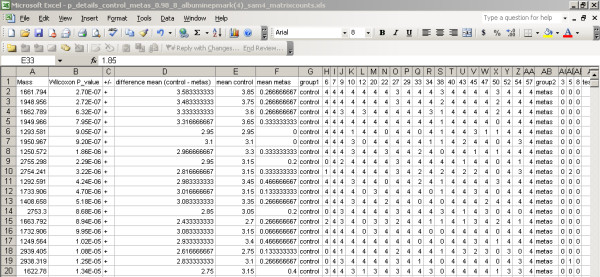
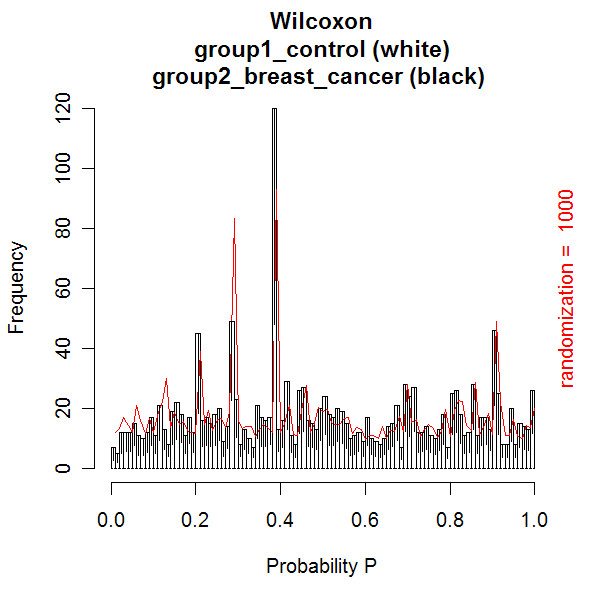
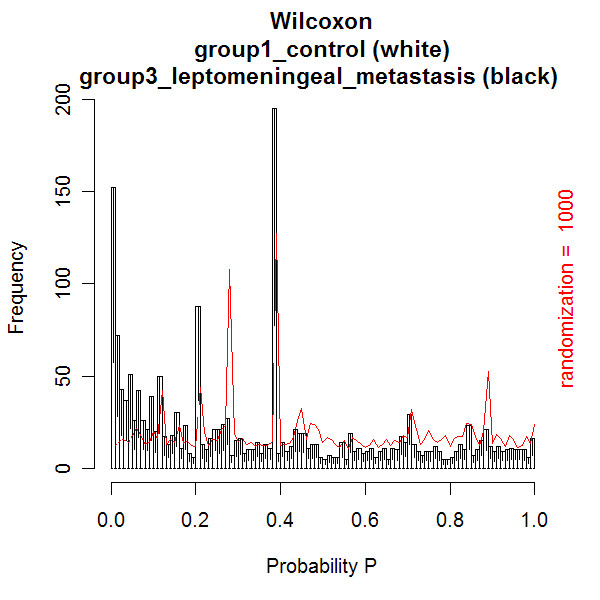
A new database application is presented, to detect and identify significantly differentially expressed peptides in peptide profiles obtained from body fluids of patient and control groups. The presented modular software is capable of central storage of mass spectra and results in fast analysis. The software architecture consists of 4 pillars, 1) a Graphical User Interface written in Java, 2) a MySQL database, which contains all metadata, such as experiment numbers and sample codes, 3) a FTP (File Transport Protocol) server to store all raw mass spectrometry files and processed data, and 4) the software package R, which is used for modular statistical calculations, such as the Wilcoxon-Mann-Whitney rank sum test. Statistic analysis by the Wilcoxon-Mann-Whitney test in R demonstrates that peptide-profiles of two patient groups 1) breast cancer patients with leptomeningeal metastases and 2) prostate cancer patients in end stage disease can be distinguished from those of control groups.
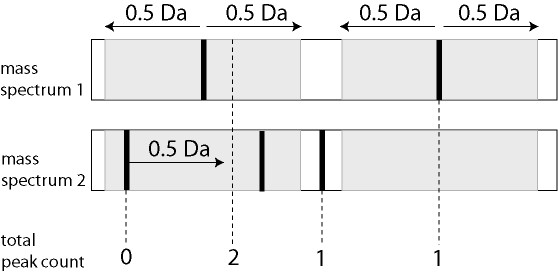
The database application is capable to distinguish patient Matrix Assisted Laser Desorption Ionization (MALDI-TOF) peptide profiles from control groups using large size datasets. The modular architecture of the application makes it possible to adapt the application to handle also large sized data from MS/MS- and Fourier Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometry experiments. It is expected that the higher resolution and mass accuracy of the FT-ICR mass spectrometry prevents the clustering of peaks of different peptides and allows the identification of differentially expressed proteins from the peptide profiles.
在生物标志物发现中对肽谱进行统计比较需要快速、用户友好的软件来进行高通量数据分析。重要特性包括在更改输入变量方面的灵活性以及对患者组和对照组之间差异表达肽的统计分析。此外,将质谱数据与其他实验结果(如微阵列分析)以及来自其他数据库的信息进行整合,需要肽谱矩阵的中央存储,在其中可以将蛋白质标识符添加到感兴趣的肽质量上。
提出了一种新的数据库应用程序,用于检测和识别从患者和对照组体液中获得的肽谱中显著差异表达的肽。所展示的模块化软件能够对质谱进行中央存储并实现快速分析。软件架构由4个支柱组成:1)用Java编写的图形用户界面;2)MySQL数据库,其中包含所有元数据,如实验编号和样本代码;3)一个FTP(文件传输协议)服务器,用于存储所有原始质谱文件和处理后的数据;4)软件包R,用于进行模块化统计计算,如Wilcoxon-Mann-Whitney秩和检验。R中通过Wilcoxon-Mann-Whitney检验进行的统计分析表明,两个患者组(1)患有软脑膜转移的乳腺癌患者和(2)终末期前列腺癌患者的肽谱可以与对照组的肽谱区分开来。
该数据库应用程序能够使用大型数据集将患者的基质辅助激光解吸电离(MALDI-TOF)肽谱与对照组区分开来。该应用程序的模块化架构使其有可能进行调整,以处理来自串联质谱(MS/MS)和傅里叶变换离子回旋共振(FT-ICR)质谱实验的大型数据。预计FT-ICR质谱的更高分辨率和质量精度可防止不同肽峰的聚集,并允许从肽谱中识别差异表达的蛋白质。