Tantoso Erwin, Yang Yuchen, Li Kuo-Bin
Bioinformatics Institute, 30 Biopolis Street, 07-01 Matrix, 138671, Singapore.
BMC Genomics. 2006 Sep 19;7:238. doi: 10.1186/1471-2164-7-238.
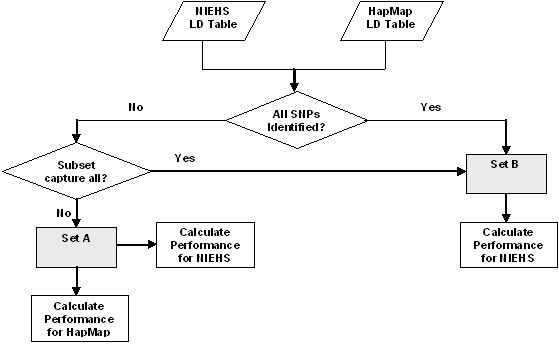
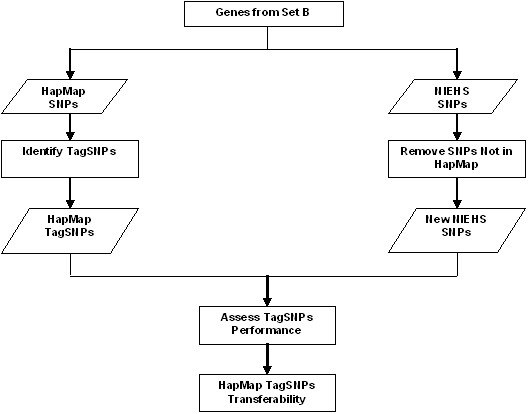
The recent advancement in human genome sequencing and genotyping has revealed millions of single nucleotide polymorphisms (SNP) which determine the variation among human beings. One of the particular important projects is The International HapMap Project which provides the catalogue of human genetic variation for disease association studies. In this paper, we analyzed the genotype data in HapMap project by using National Institute of Environmental Health Sciences Environmental Genome Project (NIEHS EGP) SNPs. We first determine whether the HapMap data are transferable to the NIEHS data. Then, we study how well the HapMap SNPs capture the untyped SNPs in the region. Finally, we provide general guidelines for determining whether the SNPs chosen from HapMap may be able to capture most of the untyped SNPs.
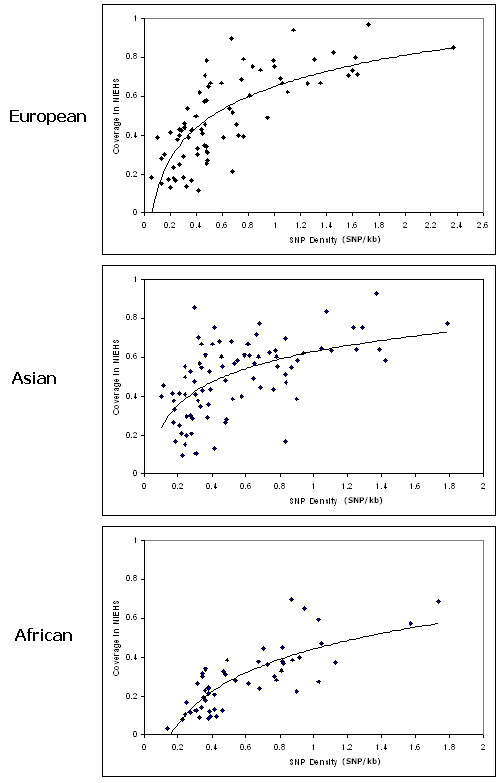
Our analysis shows that HapMap data are not robust enough to capture the untyped variants for most of the human genes. The performance of SNPs for European and Asian samples are marginal in capturing the untyped variants, i.e. approximately 55%. Expectedly, the SNPs from HapMap YRI panel can only capture approximately 30% of the variants. Although the overall performance is low, however, the SNPs for some genes perform very well and are able to capture most of the variants along the gene. This is observed in the European and Asian panel, but not in African panel. Through observation, we concluded that in order to have a well covered SNPs reference panel, the SNPs density and the association among reference SNPs are important to estimate the robustness of the chosen SNPs.
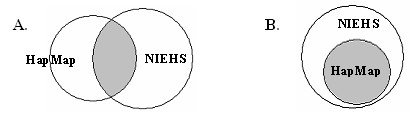
We have analyzed the coverage of HapMap SNPs using NIEHS EGP data. The results show that HapMap SNPs are transferable to the NIEHS SNPs. However, HapMap SNPs cannot capture some of the untyped SNPs and therefore resequencing may be needed to uncover more SNPs in the missing region.
人类基因组测序和基因分型的最新进展揭示了数百万个单核苷酸多态性(SNP),这些多态性决定了人类之间的差异。其中一个特别重要的项目是国际人类基因组单体型图计划(The International HapMap Project),该计划为疾病关联研究提供了人类遗传变异目录。在本文中,我们使用美国国立环境卫生科学研究所环境基因组计划(NIEHS EGP)的SNP分析了人类基因组单体型图计划中的基因型数据。我们首先确定人类基因组单体型图计划的数据是否可转移到NIEHS数据中。然后,我们研究人类基因组单体型图计划的SNP在捕获该区域未分型SNP方面的效果如何。最后,我们提供了一些通用指南,用于确定从人类基因组单体型图计划中选择的SNP是否能够捕获大多数未分型的SNP。
我们的分析表明,对于大多数人类基因,人类基因组单体型图计划的数据在捕获未分型变异方面不够稳健。欧洲和亚洲样本的SNP在捕获未分型变异方面表现一般,即大约为55%。不出所料,人类基因组单体型图计划中来自YRI群体的SNP只能捕获大约30%的变异。尽管总体表现不佳,但是,某些基因的SNP表现非常好,能够捕获该基因上的大多数变异。在欧洲和亚洲样本中观察到了这种情况,但在非洲样本中未观察到。通过观察,我们得出结论,为了有一个覆盖良好的SNP参考面板,SNP密度以及参考SNP之间的关联性对于评估所选SNP的稳健性很重要。
我们使用NIEHS EGP数据分析了人类基因组单体型图计划SNP的覆盖范围。结果表明,人类基因组单体型图计划的SNP可转移到NIEHS的SNP中。然而,人类基因组单体型图计划的SNP无法捕获一些未分型的SNP,因此可能需要重新测序以发现缺失区域中更多的SNP。