Song Jiangning, Burrage Kevin
Advanced Computational Modelling Centre, The University of Queensland, Brisbane Qld 4072, Australia.
BMC Bioinformatics. 2006 Oct 3;7:425. doi: 10.1186/1471-2105-7-425.
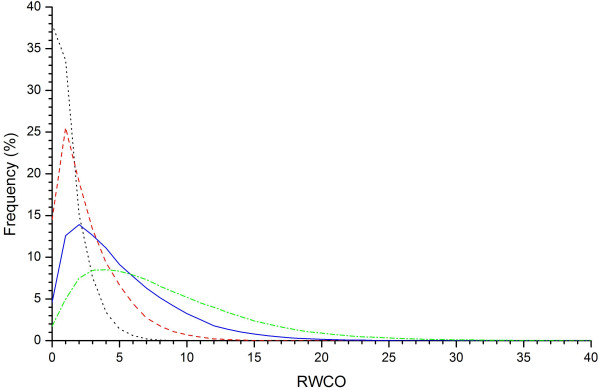
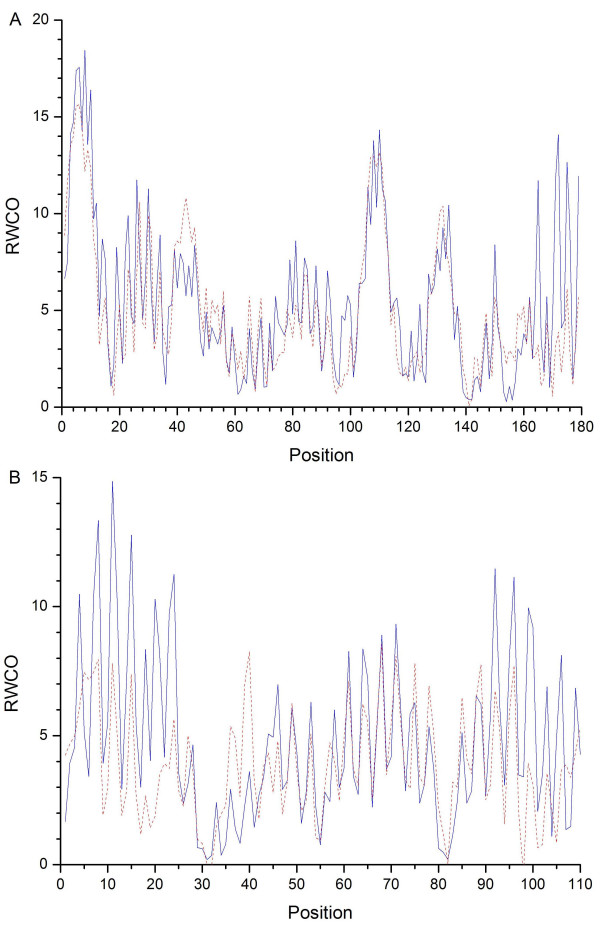
The residue-wise contact order (RWCO) describes the sequence separations between the residues of interest and its contacting residues in a protein sequence. It is a new kind of one-dimensional protein structure that represents the extent of long-range contacts and is considered as a generalization of contact order. Together with secondary structure, accessible surface area, the B factor, and contact number, RWCO provides comprehensive and indispensable important information to reconstructing the protein three-dimensional structure from a set of one-dimensional structural properties. Accurately predicting RWCO values could have many important applications in protein three-dimensional structure prediction and protein folding rate prediction, and give deep insights into protein sequence-structure relationships.
We developed a novel approach to predict residue-wise contact order values in proteins based on support vector regression (SVR), starting from primary amino acid sequences. We explored seven different sequence encoding schemes to examine their effects on the prediction performance, including local sequence in the form of PSI-BLAST profiles, local sequence plus amino acid composition, local sequence plus molecular weight, local sequence plus secondary structure predicted by PSIPRED, local sequence plus molecular weight and amino acid composition, local sequence plus molecular weight and predicted secondary structure, and local sequence plus molecular weight, amino acid composition and predicted secondary structure. When using local sequences with multiple sequence alignments in the form of PSI-BLAST profiles, we could predict the RWCO distribution with a Pearson correlation coefficient (CC) between the predicted and observed RWCO values of 0.55, and root mean square error (RMSE) of 0.82, based on a well-defined dataset with 680 protein sequences. Moreover, by incorporating global features such as molecular weight and amino acid composition we could further improve the prediction performance with the CC to 0.57 and an RMSE of 0.79. In addition, combining the predicted secondary structure by PSIPRED was found to significantly improve the prediction performance and could yield the best prediction accuracy with a CC of 0.60 and RMSE of 0.78, which provided at least comparable performance compared with the other existing methods.
The SVR method shows a prediction performance competitive with or at least comparable to the previously developed linear regression-based methods for predicting RWCO values. In contrast to support vector classification (SVC), SVR is very good at estimating the raw value profiles of the samples. The successful application of the SVR approach in this study reinforces the fact that support vector regression is a powerful tool in extracting the protein sequence-structure relationship and in estimating the protein structural profiles from amino acid sequences.
残基接触序(RWCO)描述了蛋白质序列中感兴趣残基与其接触残基之间的序列间隔。它是一种新型的一维蛋白质结构,代表远程接触的程度,被视为接触序的一种推广。与二级结构、可及表面积、B因子和接触数一起,RWCO为从一组一维结构特性重建蛋白质三维结构提供了全面且不可或缺的重要信息。准确预测RWCO值在蛋白质三维结构预测和蛋白质折叠速率预测中可能有许多重要应用,并能深入洞察蛋白质序列与结构的关系。
我们开发了一种基于支持向量回归(SVR)从一级氨基酸序列预测蛋白质残基接触序值的新方法。我们探索了七种不同的序列编码方案来研究它们对预测性能的影响,包括PSI-BLAST谱形式的局部序列、局部序列加氨基酸组成、局部序列加分子量、局部序列加由PSIPRED预测的二级结构、局部序列加分子量和氨基酸组成、局部序列加分子量和预测的二级结构,以及局部序列加分子量、氨基酸组成和预测的二级结构。当使用PSI-BLAST谱形式的具有多序列比对的局部序列时,基于一个包含680个蛋白质序列的明确数据集,我们可以预测RWCO分布,预测值与观测值之间的皮尔逊相关系数(CC)为0.55,均方根误差(RMSE)为0.82。此外,通过纳入分子量和氨基酸组成等全局特征,我们可以将预测性能进一步提高,CC达到0.57,RMSE为0.79。另外,发现结合PSIPRED预测的二级结构能显著提高预测性能,并且能产生最佳预测精度,CC为0.60,RMSE为0.78,与其他现有方法相比至少具有可比的性能。
SVR方法在预测RWCO值方面表现出与先前开发的基于线性回归的方法具有竞争力或至少相当的预测性能。与支持向量分类(SVC)不同,SVR非常擅长估计样本的原始值分布。SVR方法在本研究中的成功应用强化了这样一个事实,即支持向量回归是提取蛋白质序列与结构关系以及从氨基酸序列估计蛋白质结构分布的强大工具。