Levine David M, Haynor David R, Castle John C, Stepaniants Sergey B, Pellegrini Matteo, Mao Mao, Johnson Jason M
Rosetta Inpharmatics LLC, Merck and Co, Inc, Terry Avenue North, Seattle, WA 98109, USA.
Genome Biol. 2006;7(10):R93. doi: 10.1186/gb-2006-7-10-r93. Epub 2006 Oct 17.
Interpretation of lists of genes or proteins with altered expression is a critical and time-consuming part of microarray and proteomics research, but relatively little attention has been paid to methods for extracting biological meaning from these output lists. One powerful approach is to examine the expression of predefined biological pathways and gene sets, such as metabolic and signaling pathways and macromolecular complexes. Although many methods for measuring pathway expression have been proposed, a systematic analysis of the performance of multiple methods over multiple independent data sets has not previously been reported.
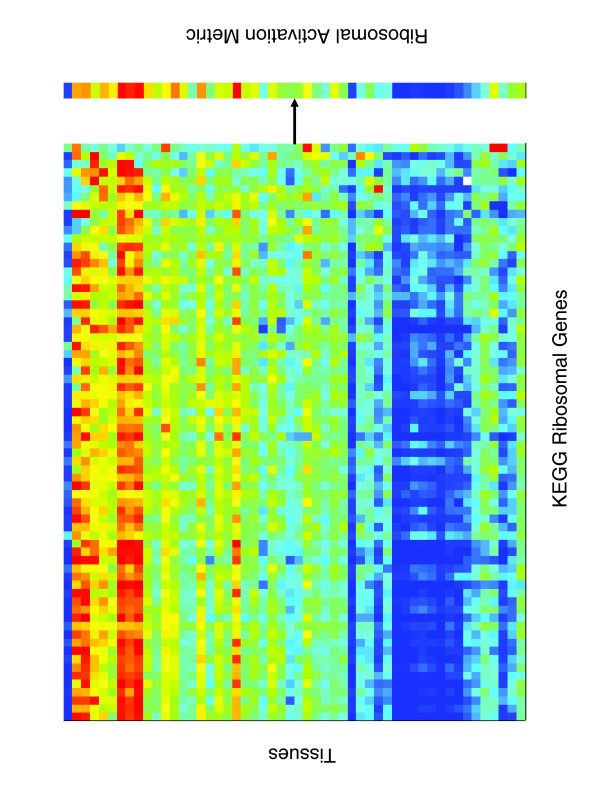
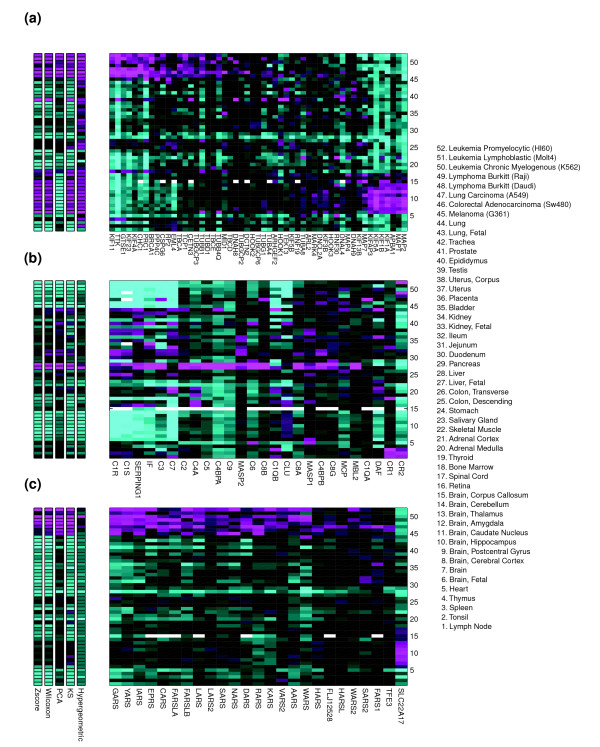
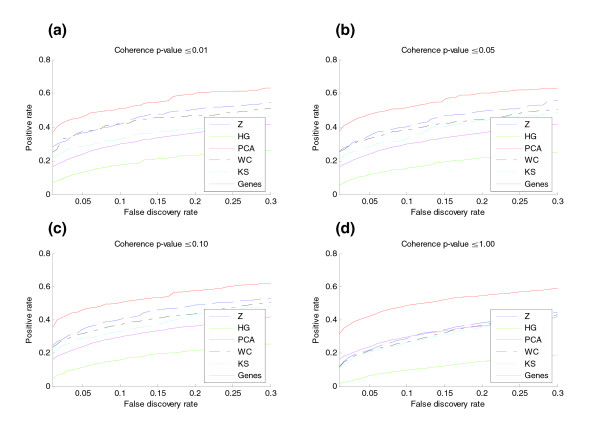
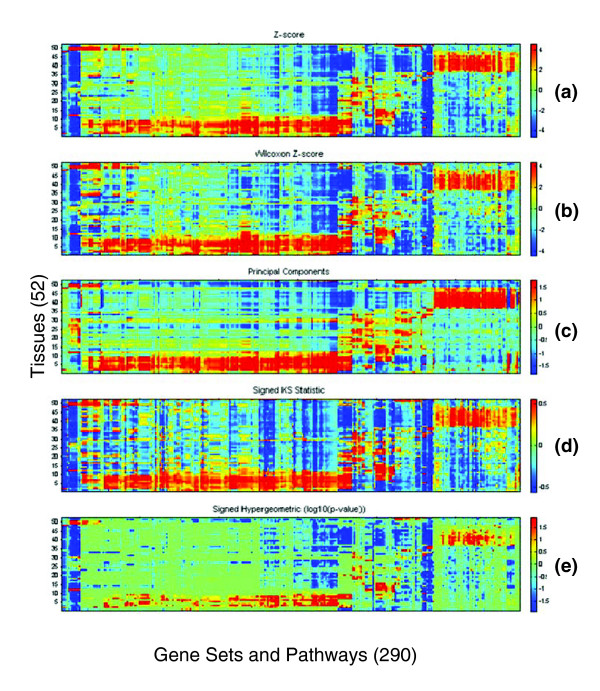
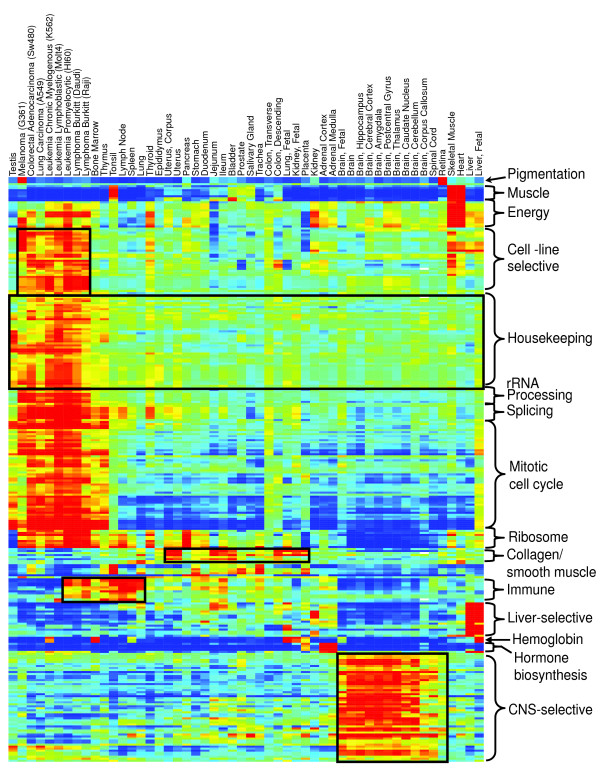
Five different measures of pathway expression were compared in an analysis of nine publicly available mRNA expression data sets. The relative sensitivity of the metrics varied greatly across data sets, and the biological pathways identified for each data set are also dependent on the choice of pathway activation metric. In addition, we show that removing incoherent pathways prior to analysis improves specificity. Finally, we create and analyze a public map of pathway expression in human tissues by gene-set analysis of a large compendium of human expression data.
We show that both the detection sensitivity and identity of pathways significantly perturbed in a microarray experiment are highly dependent on the analysis methods used and how incoherent pathways are treated. Analysts should thus consider using multiple approaches to test the robustness of their biological interpretations. We also provide a comprehensive picture of the tissue distribution of human gene pathways and a useful public archive of human pathway expression data.
解读表达发生改变的基因或蛋白质列表是微阵列和蛋白质组学研究中关键且耗时的部分,但从这些输出列表中提取生物学意义的方法却相对较少受到关注。一种有效的方法是检查预定义的生物途径和基因集的表达情况,例如代谢和信号传导途径以及大分子复合物。尽管已经提出了许多测量途径表达的方法,但此前尚未有关于在多个独立数据集上对多种方法的性能进行系统分析的报道。
在对九个公开可用的mRNA表达数据集的分析中,比较了五种不同的途径表达测量方法。这些指标的相对敏感性在不同数据集之间差异很大,并且为每个数据集确定的生物途径也取决于途径激活指标的选择。此外,我们表明在分析之前去除不连贯的途径可提高特异性。最后,我们通过对大量人类表达数据的基因集分析,创建并分析了人类组织中途径表达的公共图谱。
我们表明,在微阵列实验中显著受干扰的途径的检测敏感性和识别都高度依赖于所使用的分析方法以及如何处理不连贯的途径。因此,分析人员应考虑使用多种方法来测试其生物学解释的稳健性。我们还提供了人类基因途径组织分布的全面情况以及人类途径表达数据的有用公共存档。