Shedden Kerby, Chen Wei, Kuick Rork, Ghosh Debashis, Macdonald James, Cho Kathleen R, Giordano Thomas J, Gruber Stephen B, Fearon Eric R, Taylor Jeremy M G, Hanash Samir
Department of Statistics, University of Michigan, Ann Arbor, Michigan, USA.
BMC Bioinformatics. 2005 Feb 10;6:26. doi: 10.1186/1471-2105-6-26.
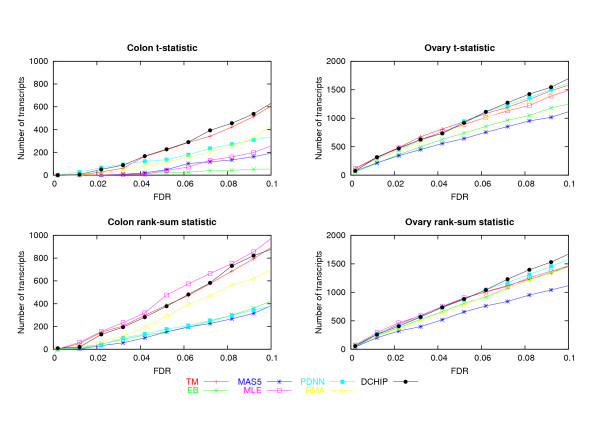
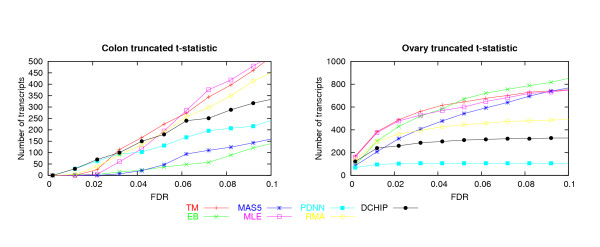
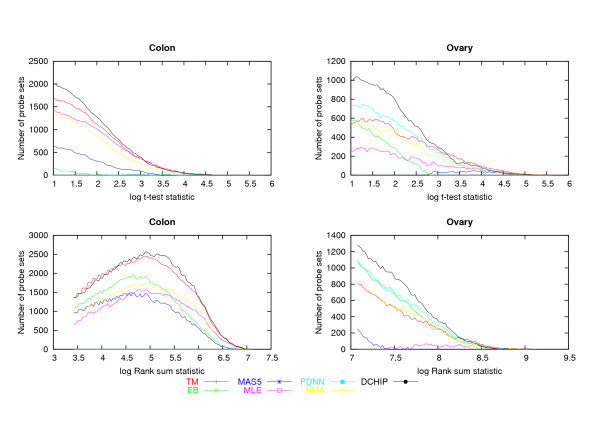
A critical step in processing oligonucleotide microarray data is combining the information in multiple probes to produce a single number that best captures the expression level of a RNA transcript. Several systematic studies comparing multiple methods for array processing have used tightly controlled calibration data sets as the basis for comparison. Here we compare performances for seven processing methods using two data sets originally collected for disease profiling studies. An emphasis is placed on understanding sensitivity for detecting differentially expressed genes in terms of two key statistical determinants: test statistic variability for non-differentially expressed genes, and test statistic size for truly differentially expressed genes.
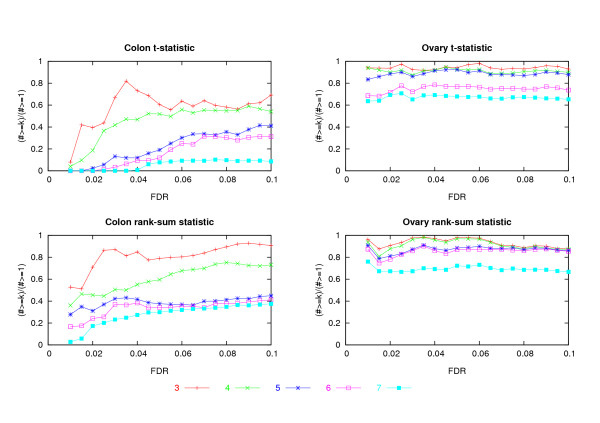
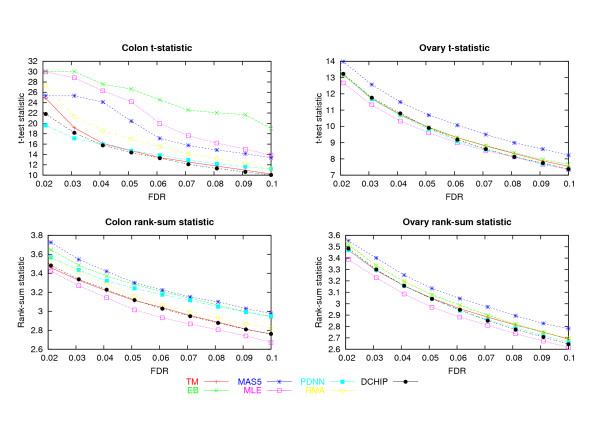
In the two data sets considered here, up to seven-fold variation across the processing methods was found in the number of genes detected at a given false discovery rate (FDR). The best performing methods called up to 90% of the same genes differentially expressed, had less variable test statistics under randomization, and had a greater number of large test statistics in the experimental data. Poor performance of one method was directly tied to a tendency to produce highly variable test statistic values under randomization. Based on an overall measure of performance, two of the seven methods (Dchip and a trimmed mean approach) are superior in the two data sets considered here. Two other methods (MAS5 and GCRMA-EB) are inferior, while results for the other three methods are mixed.
Choice of processing method has a major impact on differential expression analysis of microarray data. Previously reported performance analyses using tightly controlled calibration data sets are not highly consistent with results reported here using data from human tissue samples. Performance of array processing methods in disease profiling and other realistic biological studies should be given greater consideration when comparing Affymetrix processing methods.
处理寡核苷酸微阵列数据的关键步骤是整合多个探针中的信息,以生成一个最能反映RNA转录本表达水平的单一数值。多项比较多种阵列处理方法的系统研究使用了严格控制的校准数据集作为比较基础。在此,我们使用最初为疾病谱研究收集的两个数据集,比较了七种处理方法的性能。重点在于从两个关键统计决定因素方面理解检测差异表达基因的灵敏度:非差异表达基因的检验统计量变异性,以及真正差异表达基因的检验统计量大小。
在此处考虑的两个数据集中,发现在给定错误发现率(FDR)下检测到的基因数量,各处理方法之间存在高达七倍的差异。表现最佳的方法能检测出高达90%的相同差异表达基因,在随机化情况下检验统计量的变异性较小,并且在实验数据中有更多较大的检验统计量。一种方法的不佳表现直接与在随机化情况下产生高度可变的检验统计量值的倾向相关。基于整体性能衡量,七种方法中的两种(Dchip和一种截尾均值方法)在此处考虑的两个数据集中表现更优。另外两种方法(MAS5和GCRMA - EB)表现较差,而其他三种方法的结果则好坏参半。
处理方法的选择对微阵列数据的差异表达分析有重大影响。先前使用严格控制的校准数据集进行的性能分析,与此处使用人类组织样本数据报告的结果并非高度一致。在比较Affymetrix处理方法时,应更多考虑阵列处理方法在疾病谱分析和其他实际生物学研究中的性能。