Moake Joel L
Medical Hematology Section The Methodist Hospital, Houston, Texas 77030, USA.
Trans Am Clin Climatol Assoc. 2004;115:201-19.
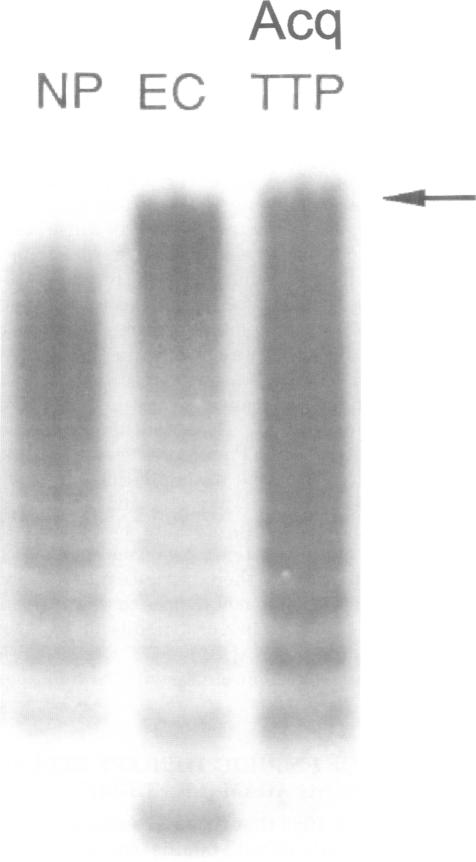
A teenager died suddenly in 1923 of systemic microvascular thrombosis. Dr. Eli Moschcowitz attributed the "hitherto undescribed disease" (now "thrombotic thrombocytopenic purpura," or "TTP") to "some powerful poison" with "both agglutinative and hemolytic properties." In 1982, TTP was found to be a defect in the "processing" of unusually large (UL) von Willebrand factor (VWF) multimers. By 1998, the cause of TTP was known to be either familial absence or acquired inhibition (by autoantibody) of plasma VWF-cleaving metalloprotease. This enzyme, the 13th member of a disintegrin and metalloprotease family with thrombospondin domains (ADAMTS-13), circulates in normal plasma waiting to cleave the long strings of ULVWF multimers emerging from stimulated endothelial cells. Uncleaved ULVWF multimers in TTP induce platelet adhesion and aggregation in the rapidly flowing blood of microvessels. Episodes of TTP are treated by "giving A DAM" (TS-13, that is) contained in normal plasma, either by infusion alone or in combination with plasmapheresis.
1923年,一名青少年因系统性微血管血栓形成突然死亡。伊莱·莫施科维茨医生将这种“此前未曾描述过的疾病”(现在称为“血栓性血小板减少性紫癜”,即“TTP”)归因于“某种具有凝集和溶血特性的强大毒素”。1982年,人们发现TTP是异常大的(UL)血管性血友病因子(VWF)多聚体“加工”过程中的缺陷。到1998年,已知TTP的病因是血浆VWF裂解金属蛋白酶的家族性缺失或获得性抑制(通过自身抗体)。这种酶是具有血小板反应蛋白结构域的去整合素和金属蛋白酶家族(ADAMTS - 13)的第13个成员,在正常血浆中循环,等待裂解从受刺激的内皮细胞中释放出来的长链ULVWF多聚体。TTP患者体内未裂解的ULVWF多聚体在微血管快速流动的血液中诱导血小板黏附和聚集。TTP发作时通过输注正常血浆中所含的“给予ADAM”(即TS - 13)进行治疗,单独输注或与血浆置换联合使用。