Kultima Kim, Scholz Birger, Alm Henrik, Sköld Karl, Svensson Marcus, Crossman Alan R, Bezard Erwan, Andrén Per E, Lönnstedt Ingrid
Department of Pharmaceutical Biosciences, Division of Toxicology, Uppsala University, BMC, Box 594, SE-75124 Uppsala, Sweden.
BMC Bioinformatics. 2006 Oct 26;7:475. doi: 10.1186/1471-2105-7-475.
Two-Dimensional Difference In Gel Electrophoresis (2D-DIGE) is a powerful tool for measuring differences in protein expression between samples or conditions. However, to remove systematic variability within and between gels the data has to be normalized. In this study we examined the ability of four existing and four novel normalization methods to remove systematic bias in data produced with 2D-DIGE. We also propose a modification of an existing method where the statistical framework determines whether a set of proteins shows an association with the predefined phenotypes of interest. This method was applied to our data generated from a monkey model (Macaca fascicularis) of Parkinson's disease.
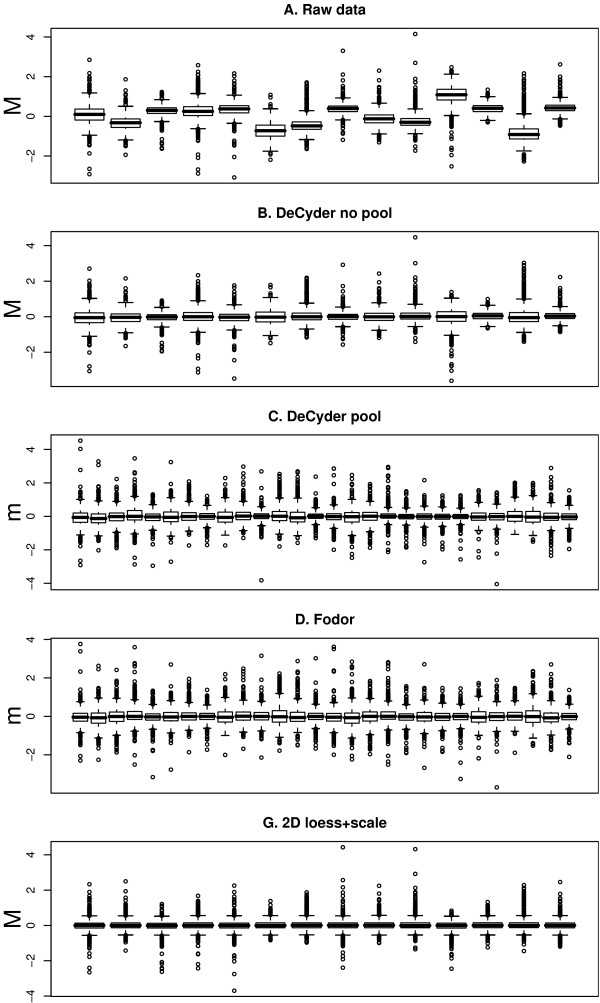
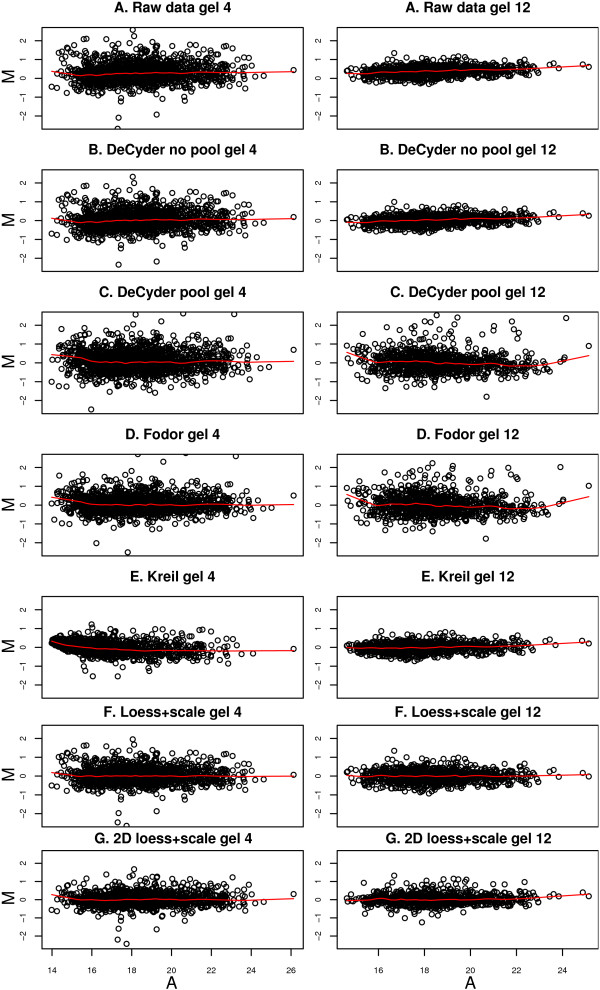
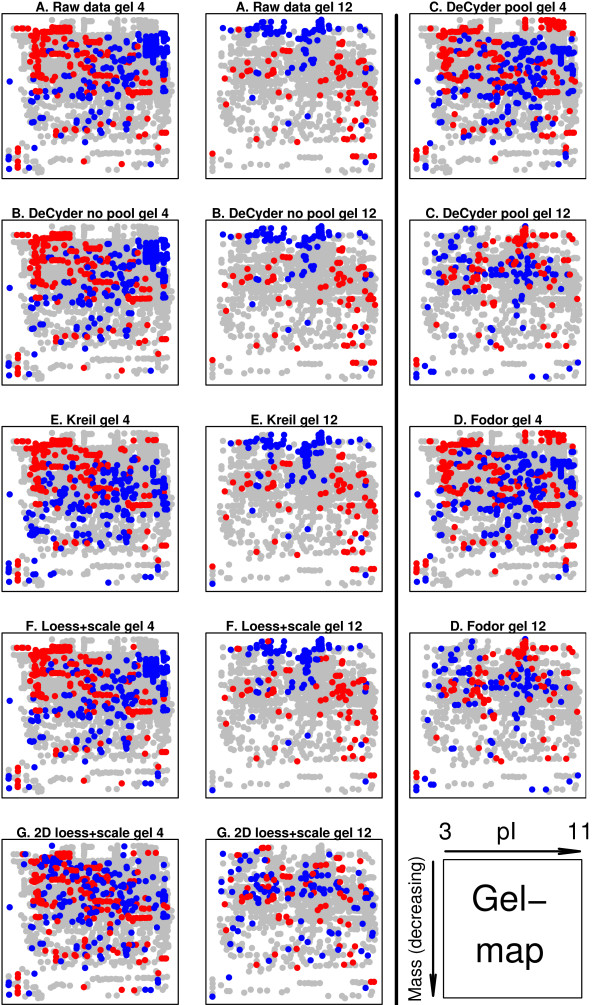
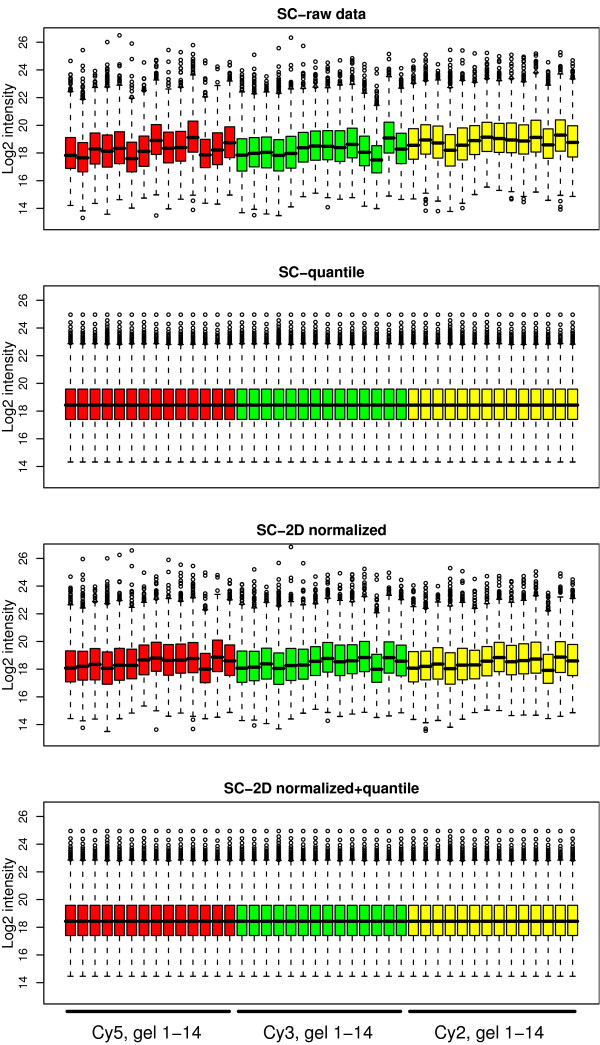
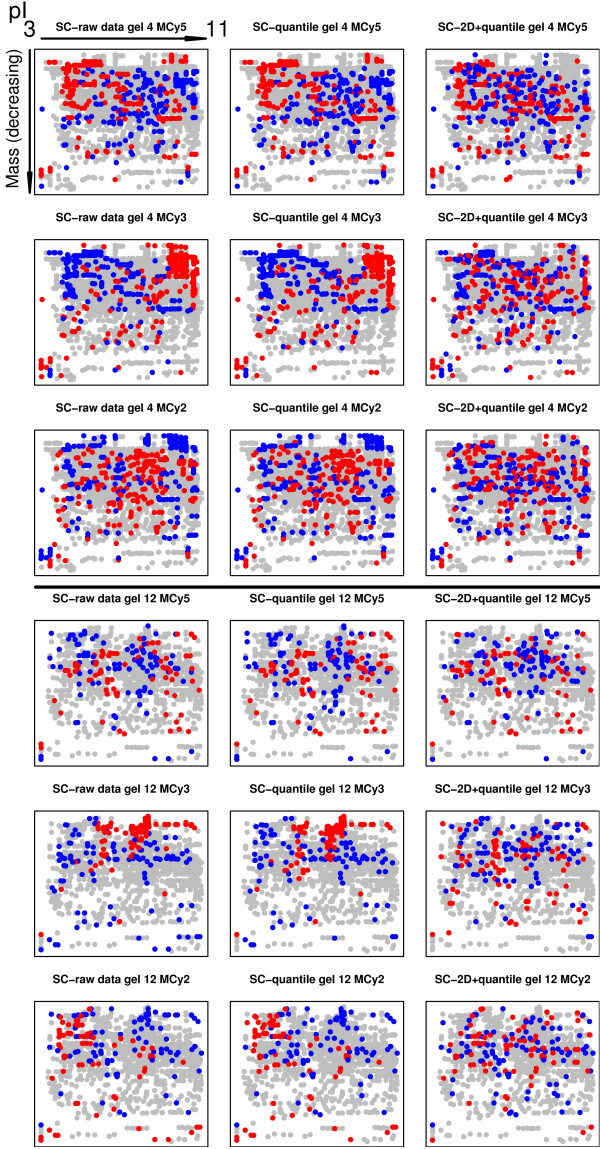
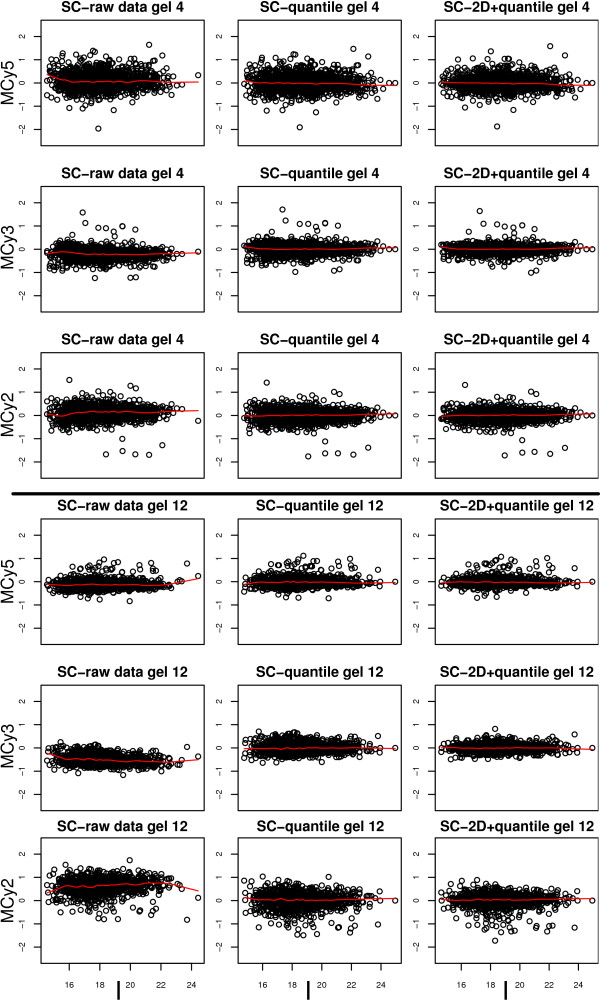
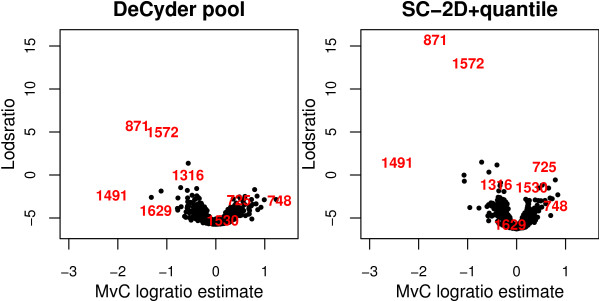
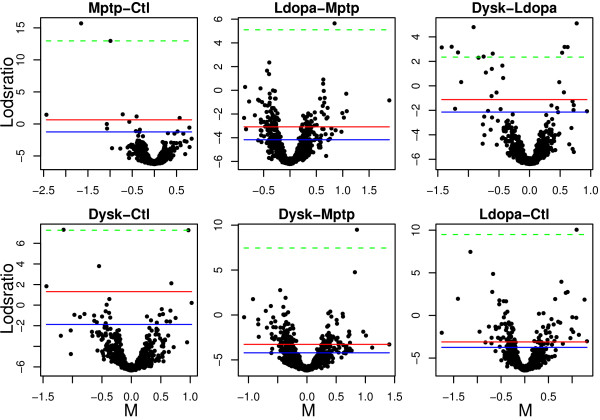
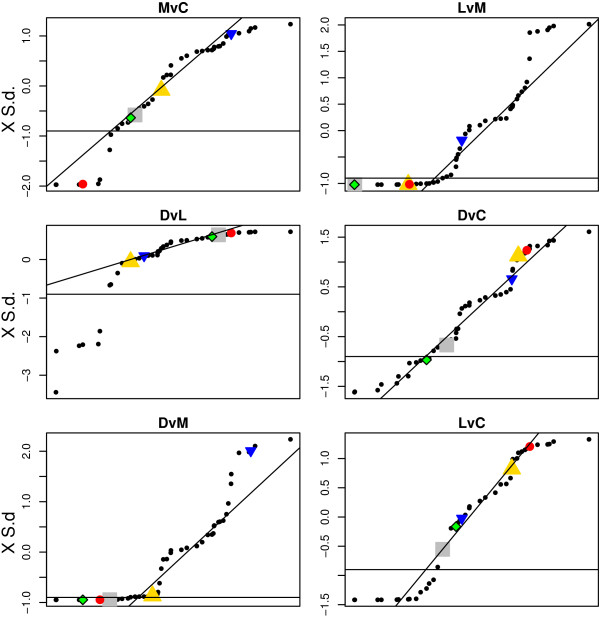
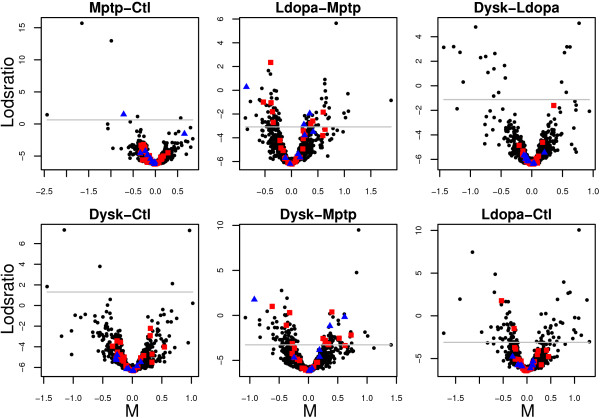
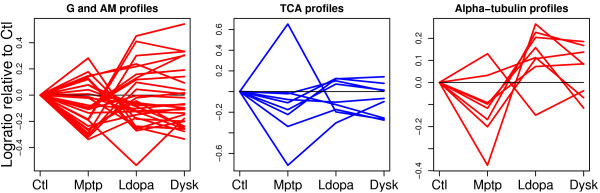
Using 2D-DIGE we analysed the protein content of the striatum from 6 control and 21 MPTP-treated monkeys, with or without de novo or long-term L-DOPA administration. There was an intensity and spatial bias in the data of all the gels examined in this study. Only two of the eight normalization methods evaluated ('2D loess+scale' and 'SC-2D+quantile') successfully removed both the intensity and spatial bias. In 'SC-2D+quantile' we extended the commonly used loess normalization method against dye bias in two-channel microarray systems to suit systems with three or more channels.Further, by using the proposed method, Differential Expression in Predefined Proteins Sets (DEPPS), several sets of proteins associated with the priming effects of L-DOPA in the striatum in parkinsonian animals were identified. Three of these sets are proteins involved in energy metabolism and one set involved proteins which are part of the microtubule cytoskeleton.
Comparison of the different methods leads to a series of methodological recommendations for the normalization and the analysis of data, depending on the experimental design. Due to the nature of 2D-DIGE data we recommend that the p-values obtained in significance tests should be used as rankings only. Individual proteins may be interesting as such, but by studying sets of proteins the interpretation of the results are probably more accurate and biologically informative.
二维差异凝胶电泳(2D-DIGE)是用于测量样本或条件之间蛋白质表达差异的强大工具。然而,为了消除凝胶内部和之间的系统变异性,数据必须进行归一化处理。在本研究中,我们检验了四种现有和四种新型归一化方法消除2D-DIGE产生的数据中系统偏差的能力。我们还提出了对现有方法的一种改进,其中统计框架确定一组蛋白质是否与感兴趣的预定义表型相关联。该方法应用于我们从帕金森病猕猴模型(食蟹猴)生成的数据。
使用2D-DIGE,我们分析了6只对照猴和21只经MPTP处理的猴纹状体的蛋白质含量,这些猴接受或未接受从头或长期左旋多巴给药。在本研究中检测的所有凝胶数据中存在强度和空间偏差。评估的八种归一化方法中只有两种(“二维局部加权回归+缩放”和“SC-2D+分位数”)成功消除了强度和空间偏差。在“SC-2D+分位数”中,我们将常用于消除双通道微阵列系统中染料偏差的局部加权回归归一化方法扩展到适用于具有三个或更多通道的系统。此外,通过使用所提出的方法,即预定义蛋白质组中的差异表达(DEPPS),鉴定了与帕金森病动物纹状体中左旋多巴引发效应相关的几组蛋白质。其中三组是参与能量代谢的蛋白质,一组是参与微管细胞骨架的蛋白质。
根据实验设计,不同方法的比较产生了一系列关于数据归一化和分析的方法学建议。由于2D-DIGE数据的性质,我们建议在显著性检验中获得的p值仅用作排名。单个蛋白质本身可能很有趣,但通过研究蛋白质组,结果的解释可能更准确且具有生物学信息性。