Ahmed Niyaz, Devi S Manjulata, Valverde M de los A, Vijayachari P, Machang'u Robert S, Ellis William A, Hartskeerl Rudy A
Pathogen Evolution Group, Centre for DNA Fingerprinting and Diagnostics, Hyderabad 500076, India.
Ann Clin Microbiol Antimicrob. 2006 Nov 23;5:28. doi: 10.1186/1476-0711-5-28.
Leptospira are the parasitic bacterial organisms associated with a broad range of mammalian hosts and are responsible for severe cases of human Leptospirosis. The epidemiology of leptospirosis is complex and dynamic. Multiple serovars have been identified, each adapted to one or more animal hosts. Adaptation is a dynamic process that changes the spatial and temporal distribution of serovars and clinical manifestations in different hosts. Serotyping based on repertoire of surface antigens is an ambiguous and artificial system of classification of leptospiral agents. Molecular typing methods for the identification of pathogenic leptospires up to individual genome species level have been highly sought after since the decipherment of whole genome sequences. Only a few resources exist for microbial genotypic data based on individual techniques such as Multiple Locus Sequence Typing (MLST), but unfortunately no such databases are existent for leptospires.
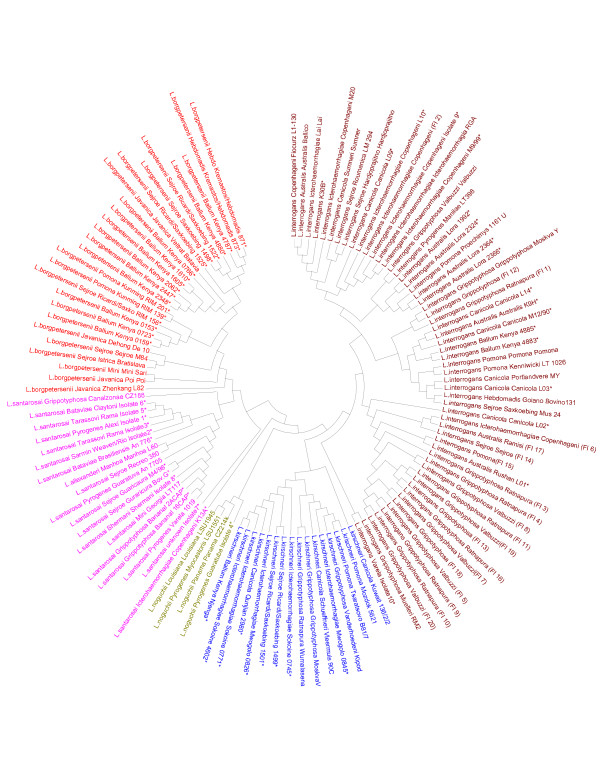
We for the first time report development of a robust MLST method for genotyping of Leptospira. Genotyping based on DNA sequence identity of 4 housekeeping genes and 2 candidate genes was analyzed in a set of 120 strains including 41 reference strains representing different geographical areas and from different sources. Of the six selected genes, adk, icdA and secY were significantly more variable whereas the LipL32 and LipL41 coding genes and the rrs2 gene were moderately variable. The phylogenetic tree clustered the isolates according to the genome-based species.
The main advantages of MLST over other typing methods for leptospires include reproducibility, robustness, consistency and portability. The genetic relatedness of the leptospires can be better studied by the MLST approach and can be used for molecular epidemiological and evolutionary studies and population genetics.
钩端螺旋体是与多种哺乳动物宿主相关的寄生细菌生物,可导致人类严重的钩端螺旋体病。钩端螺旋体病的流行病学复杂且动态变化。已鉴定出多个血清型,每个血清型适应一种或多种动物宿主。适应是一个动态过程,会改变血清型在不同宿主中的时空分布以及临床表现。基于表面抗原库的血清分型是一种模糊且人为的钩端螺旋体分类系统。自从全基因组序列被破译以来,人们一直在高度寻求用于将致病性钩端螺旋体鉴定到单个基因组物种水平的分子分型方法。基于诸如多位点序列分型(MLST)等个别技术的微生物基因型数据资源很少,不幸的是,目前尚无针对钩端螺旋体的此类数据库。
我们首次报告了一种用于钩端螺旋体基因分型的可靠MLST方法的开发。在一组120株菌株中分析了基于4个管家基因和2个候选基因的DNA序列同一性的基因分型,其中包括41株代表不同地理区域和不同来源的参考菌株。在所选的6个基因中,adk、icdA和secY的变异性明显更高,而LipL32和LipL41编码基因以及rrs2基因的变异性中等。系统发育树根据基于基因组的物种对分离株进行了聚类。
与其他钩端螺旋体分型方法相比,MLST的主要优点包括可重复性、稳健性、一致性和便携性。通过MLST方法可以更好地研究钩端螺旋体的遗传相关性,并且可用于分子流行病学、进化研究和群体遗传学。