Strop Pavel, Brzustowicz Michael R, Brunger Axel T
Howard Hughes Medical Institute and Department of Molecular and Cellular Physiology, and Stanford Synchrotron Radiation Laboratory, Stanford University, James H. Clark Center E300, Stanford, California 94305, USA.
Acta Crystallogr D Biol Crystallogr. 2007 Feb;63(Pt 2):188-96. doi: 10.1107/S0907444906045793. Epub 2007 Jan 16.
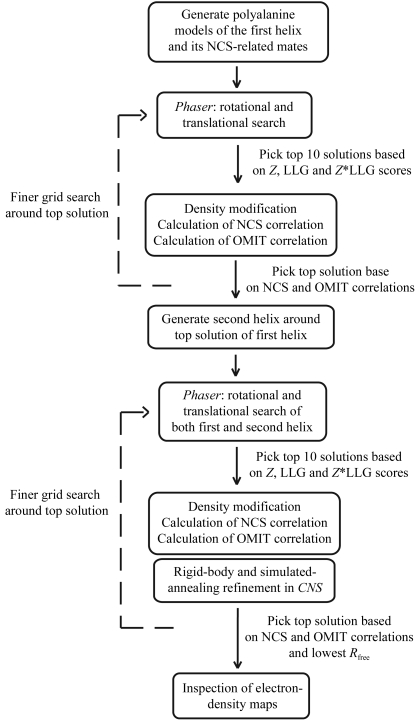
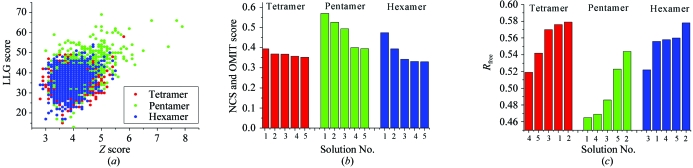
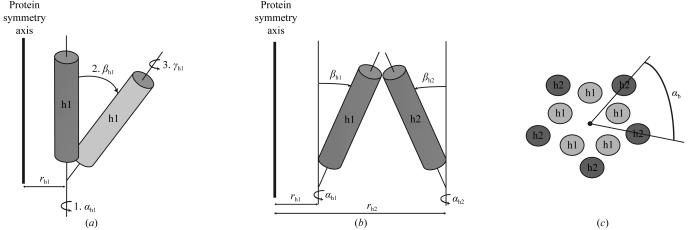
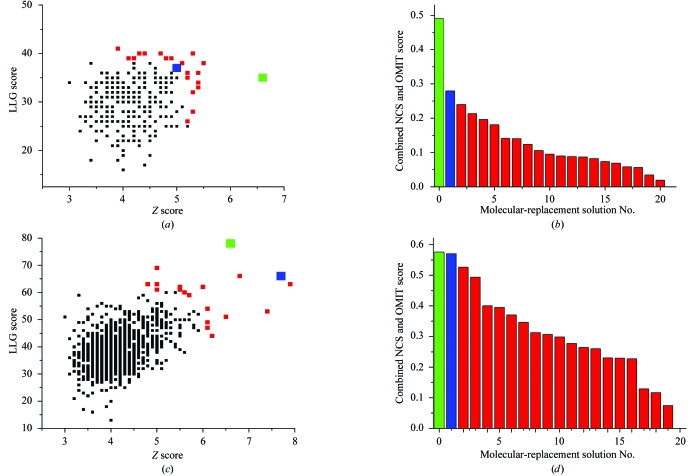
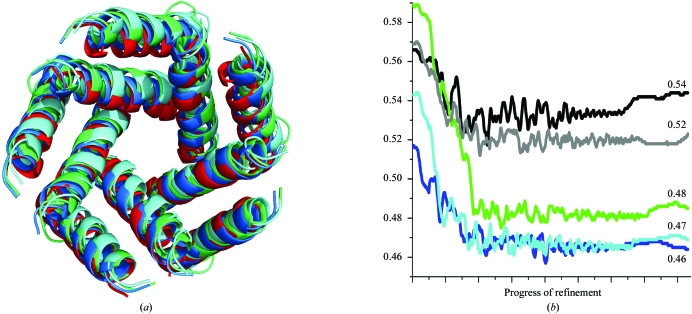
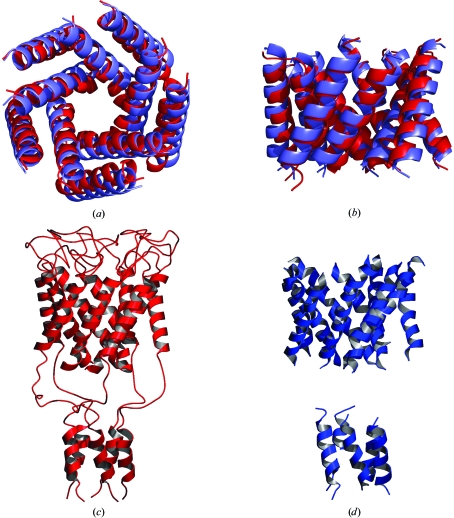
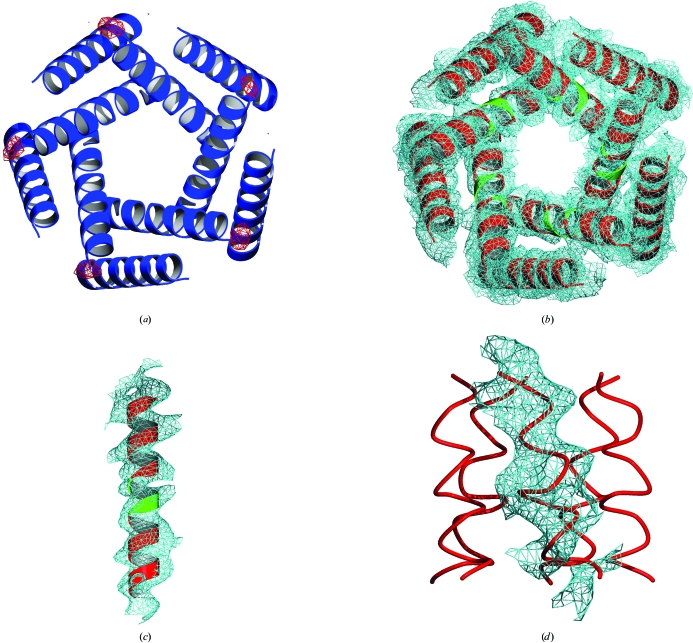
Obtaining phases for X-ray diffraction data can be a rate-limiting step in structure determination. Taking advantage of constraints specific to membrane proteins, an ab initio molecular-replacement method has been developed for phasing X-ray diffraction data for symmetric helical membrane proteins without prior knowledge of their structure or heavy-atom derivatives. The described method is based on generating all possible orientations of idealized transmembrane helices and using each model in a molecular-replacement search. The number of models is significantly reduced by taking advantage of geometrical and structural restraints specific to membrane proteins. The top molecular-replacement results are evaluated based on noncrystallographic symmetry (NCS) map correlation, OMIT map correlation and R(free) value after refinement of a polyalanine model. The feasibility of this approach is illustrated by phasing the mechanosensitive channel of large conductance (MscL) with only 4 A diffraction data. No prior structural knowledge was used other than the number of transmembrane helices. The search produced the correct spatial organization and the position in the asymmetric unit of all transmembrane helices of MscL. The resulting electron-density maps were of sufficient quality to automatically build all helical segments of MscL including the cytoplasmic domain. The method does not require high-resolution diffraction data and can be used to obtain phases for symmetrical helical membrane proteins with one or two helices per monomer.
获取X射线衍射数据的相位可能是结构测定中的一个限速步骤。利用膜蛋白特有的限制条件,已经开发出一种从头算分子置换方法,用于在没有对称螺旋膜蛋白结构或重原子衍生物先验知识的情况下确定其X射线衍射数据的相位。所描述的方法基于生成理想化跨膜螺旋的所有可能取向,并在分子置换搜索中使用每个模型。通过利用膜蛋白特有的几何和结构限制,模型数量显著减少。基于非晶体学对称性(NCS)图谱相关性、省略图谱相关性以及聚丙氨酸模型精修后的R(自由)值,对顶级分子置换结果进行评估。通过仅用4埃衍射数据对大电导机械敏感通道(MscL)进行相位测定,说明了这种方法的可行性。除了跨膜螺旋的数量外,未使用其他先验结构知识。搜索得到了MscL所有跨膜螺旋在不对称单元中的正确空间组织和位置。所得电子密度图的质量足以自动构建MscL的所有螺旋片段,包括胞质结构域。该方法不需要高分辨率衍射数据,可用于为每个单体有一个或两个螺旋的对称螺旋膜蛋白获取相位。