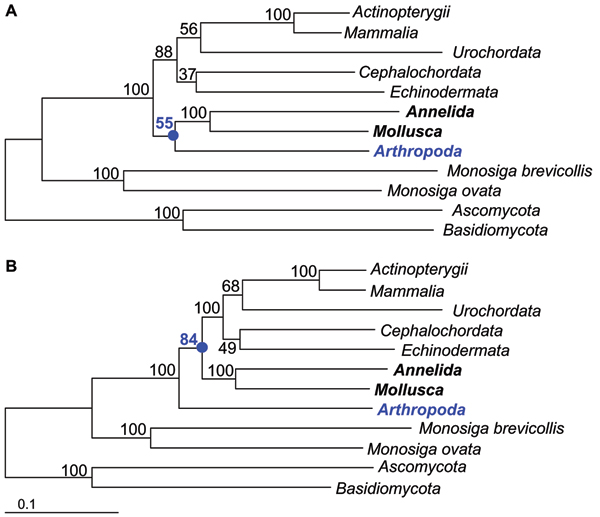
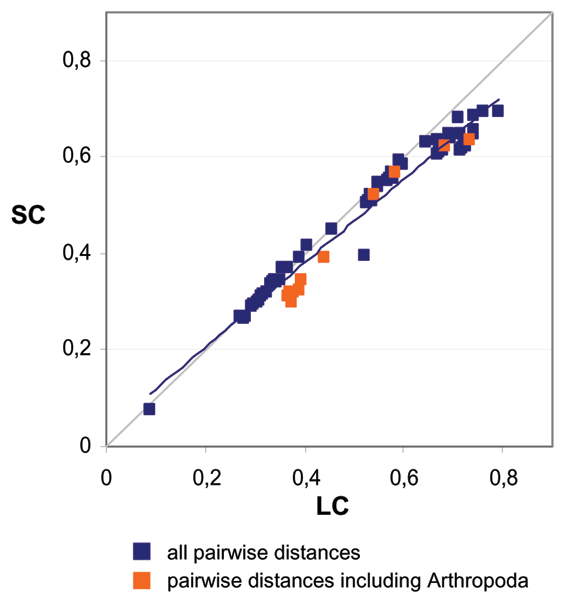
Roure Béatrice, Rodriguez-Ezpeleta Naiara, Philippe Hervé
Canadian Institute for Advanced Research, Centre Robert Cedergren, Département de biochimie, Université de Montréal, Montréal, Québec H3C3J7, Canada.
BMC Evol Biol. 2007 Feb 8;7 Suppl 1(Suppl 1):S2. doi: 10.1186/1471-2148-7-S1-S2.
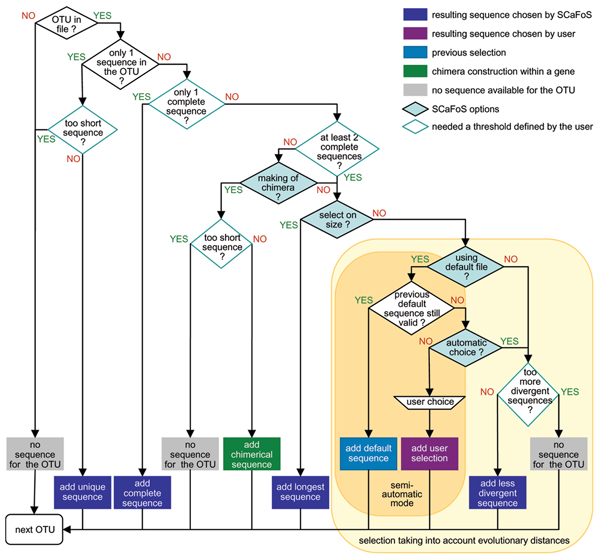
Phylogenetic analyses based on datasets rich in both genes and species (phylogenomics) are becoming a standard approach to resolve evolutionary questions. However, several difficulties are associated with the assembly of large datasets, such as multiple copies of a gene per species (paralogous or xenologous genes), lack of some genes for a given species, or partial sequences. The use of undetected paralogous or xenologous genes in phylogenetic inference can lead to inaccurate results, and the use of partial sequences to a lack of resolution. A tool that selects sequences, species, and genes, while dealing with these issues, is needed in a phylogenomics context.
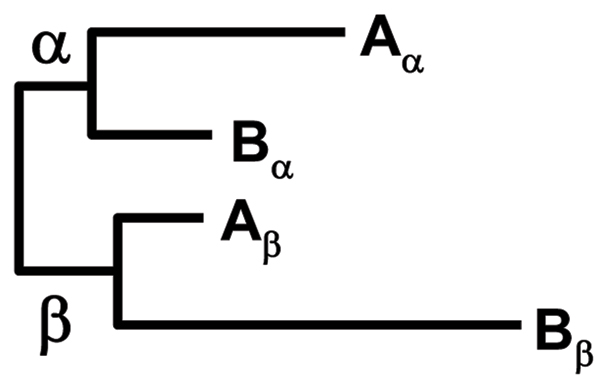
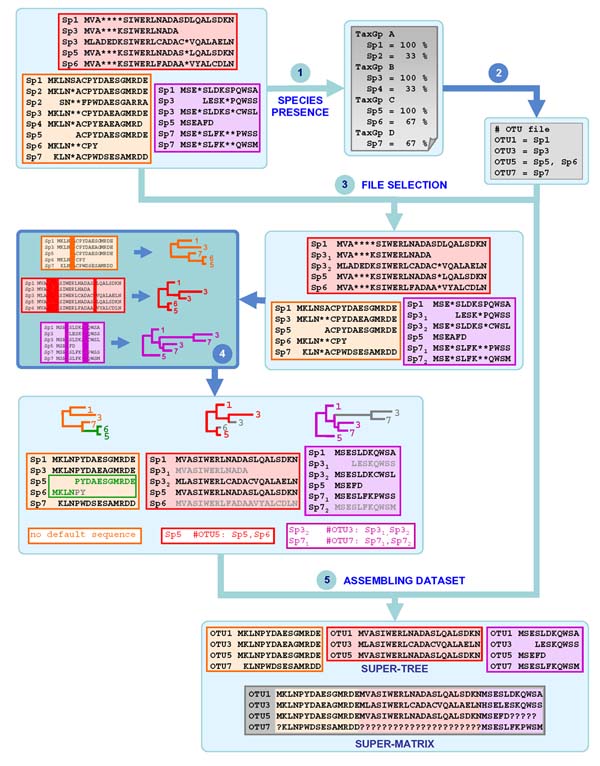
Here, we present SCaFoS, a tool that quickly assembles phylogenomic datasets containing maximal phylogenetic information while adjusting the amount of missing data in the selection of species, sequences and genes. Starting from individual sequence alignments, and using monophyletic groups defined by the user, SCaFoS creates chimeras with partial sequences, or selects, among multiple sequences, the orthologous and/or slowest evolving sequences. Once sequences representing each predefined monophyletic group have been selected, SCaFos retains genes according to the user's allowed level of missing data and generates files for super-matrix and super-tree analyses in several formats compatible with standard phylogenetic inference software. Because no clear-cut criteria exist for the sequence selection, a semi-automatic mode is available to accommodate user's expertise.
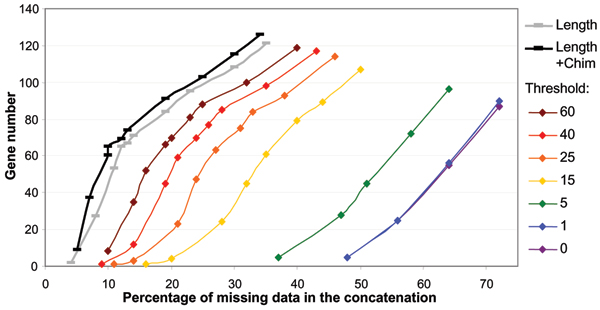
SCaFos is able to deal with datasets of hundreds of species and genes, both at the amino acid or nucleotide level. It has a graphical interface and can be integrated in an automatic workflow. Moreover, SCaFoS is the first tool that integrates user's knowledge to select orthologous sequences, creates chimerical sequences to reduce missing data and selects genes according to their level of missing data. Finally, applying SCaFoS to different datasets, we show that the judicious selection of genes, species and sequences reduces tree reconstruction artefacts, especially if the dataset includes fast evolving species.
基于富含基因和物种的数据集进行系统发育分析(系统发育基因组学)正成为解决进化问题的标准方法。然而,大型数据集的组装存在若干困难,例如每个物种的基因有多个拷贝(旁系同源或异源基因)、给定物种缺少某些基因或序列不完整。在系统发育推断中使用未检测到的旁系同源或异源基因可能导致结果不准确,而使用不完整序列则会缺乏分辨率。在系统发育基因组学背景下,需要一种能够处理这些问题的同时选择序列、物种和基因的工具。
在此,我们展示了SCaFoS,这是一种工具,它能快速组装包含最大系统发育信息的系统发育基因组数据集,同时在物种、序列和基因的选择中调整缺失数据的量。从单个序列比对开始,并使用用户定义的单系类群,SCaFoS创建具有不完整序列的嵌合体,或在多个序列中选择直系同源和/或进化最慢的序列。一旦选择了代表每个预定义单系类群的序列,SCaFos会根据用户允许的缺失数据水平保留基因,并生成与标准系统发育推断软件兼容的多种格式的超级矩阵和超级树分析文件。由于序列选择没有明确的标准,因此提供了半自动模式以适应用户的专业知识。
SCaFos能够处理数百个物种和基因的数据集,无论是氨基酸水平还是核苷酸水平。它具有图形界面,并且可以集成到自动工作流程中。此外,SCaFoS是第一个整合用户知识以选择直系同源序列、创建嵌合序列以减少缺失数据并根据其缺失数据水平选择基因的工具。最后,将SCaFoS应用于不同的数据集,我们表明明智地选择基因、物种和序列可以减少树重建假象,特别是如果数据集中包括快速进化的物种。