Schreiber Fabian, Pick Kerstin, Erpenbeck Dirk, Wörheide Gert, Morgenstern Burkhard
Abteilung Bioinformatik, Institut für Mikrobiologie und Genetik, Georg-August-Universität Göttingen, Göttingen, Germany.
BMC Bioinformatics. 2009 Jul 16;10:219. doi: 10.1186/1471-2105-10-219.
Phylogenetic studies using expressed sequence tags (EST) are becoming a standard approach to answer evolutionary questions. Such studies are usually based on large sets of newly generated, unannotated, and error-prone EST sequences from different species. A first crucial step in EST-based phylogeny reconstruction is to identify groups of orthologous sequences. From these data sets, appropriate target genes are selected, and redundant sequences are eliminated to obtain suitable sequence sets as input data for tree-reconstruction software. Generating such data sets manually can be very time consuming. Thus, software tools are needed that carry out these steps automatically.
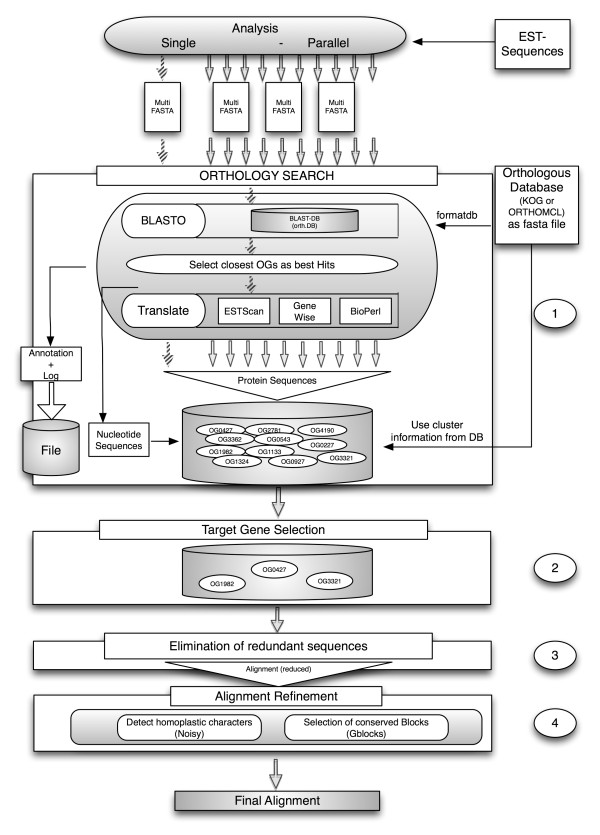
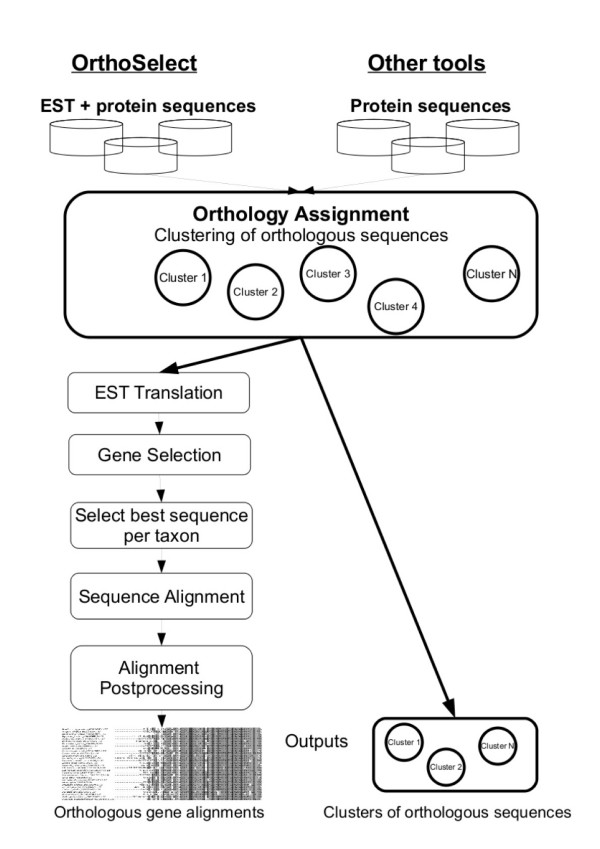
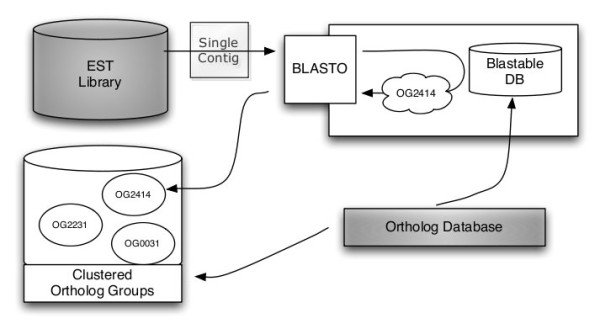
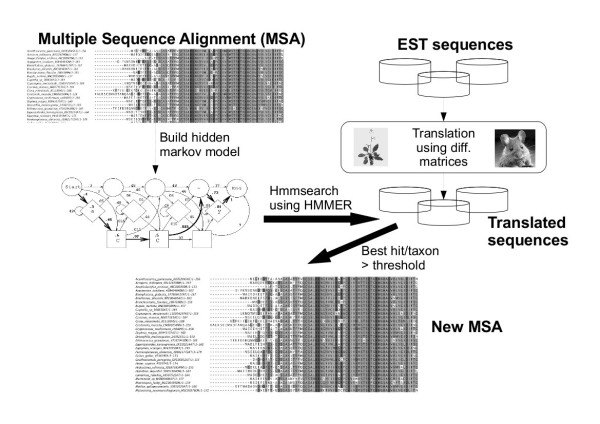
We developed a flexible and user-friendly software pipeline, running on desktop machines or computer clusters, that constructs data sets for phylogenomic analyses. It automatically searches assembled EST sequences against databases of orthologous groups (OG), assigns ESTs to these predefined OGs, translates the sequences into proteins, eliminates redundant sequences assigned to the same OG, creates multiple sequence alignments of identified orthologous sequences and offers the possibility to further process this alignment in a last step by excluding potentially homoplastic sites and selecting sufficiently conserved parts. Our software pipeline can be used as it is, but it can also be adapted by integrating additional external programs. This makes the pipeline useful for non-bioinformaticians as well as to bioinformatic experts. The software pipeline is especially designed for ESTs, but it can also handle protein sequences.
OrthoSelect is a tool that produces orthologous gene alignments from assembled ESTs. Our tests show that OrthoSelect detects orthologs in EST libraries with high accuracy. In the absence of a gold standard for orthology prediction, we compared predictions by OrthoSelect to a manually created and published phylogenomic data set. Our tool was not only able to rebuild the data set with a specificity of 98%, but it detected four percent more orthologous sequences. Furthermore, the results OrthoSelect produces are in absolut agreement with the results of other programs, but our tool offers a significant speedup and additional functionality, e.g. handling of ESTs, computing sequence alignments, and refining them. To our knowledge, there is currently no fully automated and freely available tool for this purpose. Thus, OrthoSelect is a valuable tool for researchers in the field of phylogenomics who deal with large quantities of EST sequences. OrthoSelect is written in Perl and runs on Linux/Mac OS X. The tool can be downloaded at (http://gobics.de/fabian/orthoselect.php).
利用表达序列标签(EST)进行系统发育研究正逐渐成为解答进化问题的标准方法。此类研究通常基于来自不同物种的大量新生成的、未注释且容易出错的EST序列。基于EST的系统发育重建的关键第一步是识别直系同源序列组。从这些数据集中,选择合适的目标基因,并去除冗余序列,以获得合适的序列集作为树重建软件的输入数据。手动生成此类数据集可能非常耗时。因此,需要能自动执行这些步骤的软件工具。
我们开发了一种灵活且用户友好的软件管道,可在桌面计算机或计算机集群上运行,用于构建系统发育基因组分析的数据集。它会自动针对直系同源组(OG)数据库搜索组装好的EST序列,将EST分配到这些预定义的OG中,将序列翻译成蛋白质,去除分配到同一OG的冗余序列,创建已识别直系同源序列的多序列比对,并提供在最后一步通过排除潜在的平行位点和选择足够保守的部分来进一步处理此比对的可能性。我们的软件管道可以直接使用,也可以通过集成其他外部程序进行调整。这使得该管道对非生物信息学家以及生物信息学专家都很有用。该软件管道专门为EST设计,但也可以处理蛋白质序列。
OrthoSelect是一种从组装好的EST中生成直系同源基因比对的工具。我们的测试表明,OrthoSelect能高精度地检测EST文库中的直系同源物。在缺乏直系同源预测的金标准的情况下,我们将OrthoSelect的预测结果与一个手动创建并已发表的系统发育基因组数据集进行了比较。我们的工具不仅能够以98%的特异性重建该数据集,而且还检测到了多4%的直系同源序列。此外,OrthoSelect产生的结果与其他程序的结果完全一致,但我们的工具显著加快了速度并提供了额外功能,例如处理EST、计算序列比对并对其进行优化。据我们所知,目前尚无用于此目的的完全自动化且免费可用的工具。因此,OrthoSelect对于处理大量EST序列的系统发育基因组学领域的研究人员来说是一个有价值的工具。OrthoSelect用Perl编写,可在Linux/Mac OS X上运行。该工具可从(http://gobics.de/fabian/orthoselect.php)下载。