Lartillot Nicolas, Brinkmann Henner, Philippe Hervé
Laboratoire d'Informatique, de Robotique et de Microélectronique de Montpellier, UMR 5506, CNRS-Université de Montpellier 2, Montpellier Cedex 5, France.
BMC Evol Biol. 2007 Feb 8;7 Suppl 1(Suppl 1):S4. doi: 10.1186/1471-2148-7-S1-S4.
Thanks to the large amount of signal contained in genome-wide sequence alignments, phylogenomic analyses are converging towards highly supported trees. However, high statistical support does not imply that the tree is accurate. Systematic errors, such as the Long Branch Attraction (LBA) artefact, can be misleading, in particular when the taxon sampling is poor, or the outgroup is distant. In an otherwise consistent probabilistic framework, systematic errors in genome-wide analyses can be traced back to model mis-specification problems, which suggests that better models of sequence evolution should be devised, that would be more robust to tree reconstruction artefacts, even under the most challenging conditions.
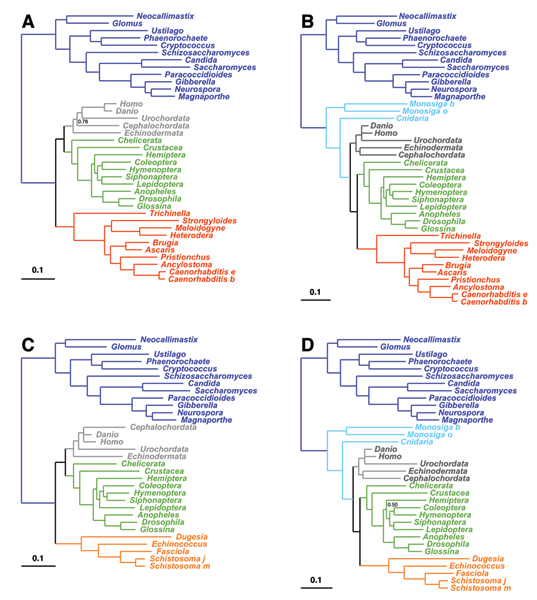
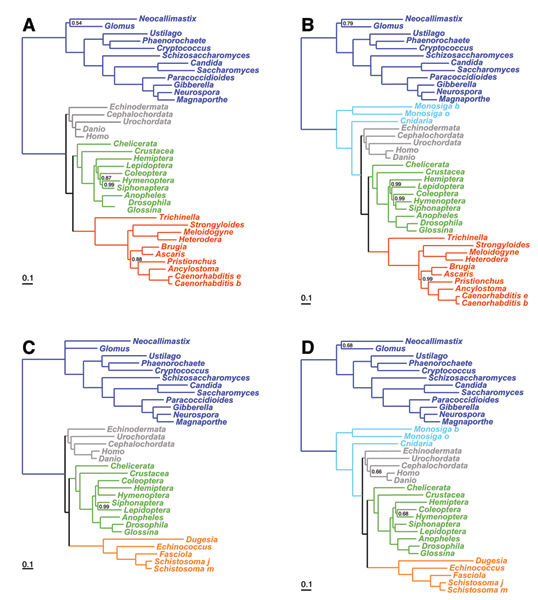
We focus on a well characterized LBA artefact analyzed in a previous phylogenomic study of the metazoan tree, in which two fast-evolving animal phyla, nematodes and platyhelminths, emerge either at the base of all other Bilateria, or within protostomes, depending on the outgroup. We use this artefactual result as a case study for comparing the robustness of two alternative models: a standard, site-homogeneous model, based on an empirical matrix of amino-acid replacement (WAG), and a site-heterogeneous mixture model (CAT). In parallel, we propose a posterior predictive test, allowing one to measure how well a model acknowledges sequence saturation.
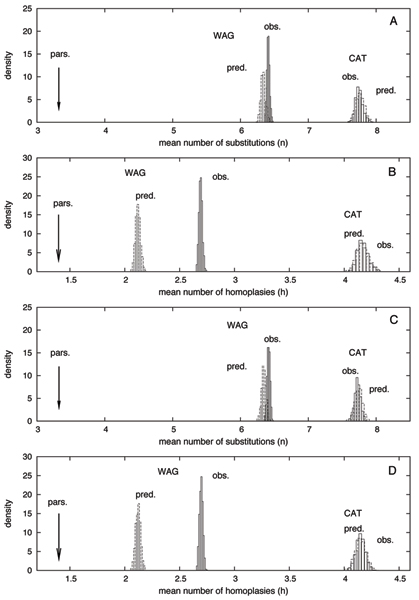
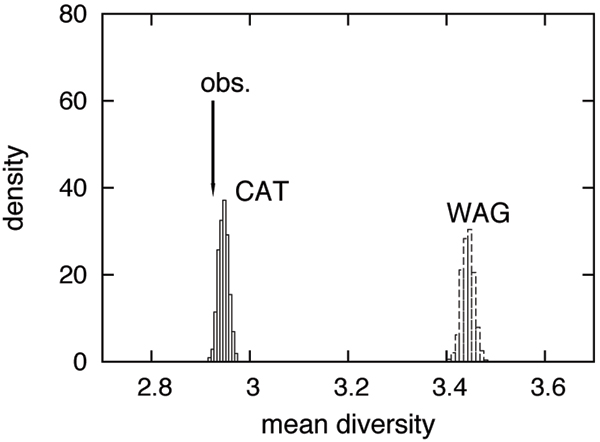
Adopting a Bayesian framework, we show that the LBA artefact observed under WAG disappears when the site-heterogeneous model CAT is used. Using cross-validation, we further demonstrate that CAT has a better statistical fit than WAG on this data set. Finally, using our statistical goodness-of-fit test, we show that CAT, but not WAG, correctly accounts for the overall level of saturation, and that this is due to a better estimation of site-specific amino-acid preferences.
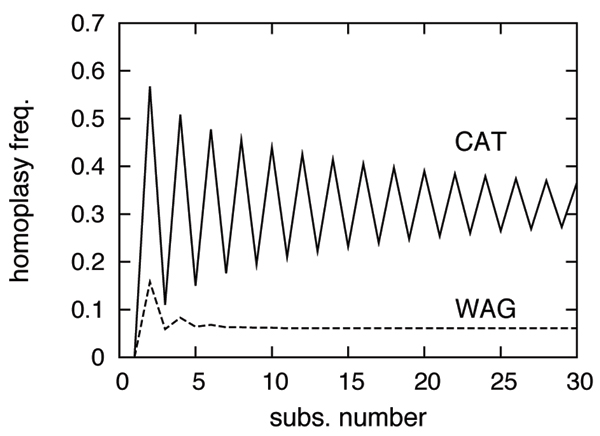
The CAT model appears to be more robust than WAG against LBA artefacts, essentially because it correctly anticipates the high probability of convergences and reversions implied by the small effective size of the amino-acid alphabet at each site of the alignment. More generally, our results provide strong evidence that site-specificities in the substitution process need be accounted for in order to obtain more reliable phylogenetic trees.
由于全基因组序列比对中包含大量信号,系统发育基因组学分析正趋向于得到高度支持的树。然而,高统计支持并不意味着树是准确的。系统误差,如长枝吸引(LBA)假象,可能会产生误导,特别是当分类群抽样不足或外类群距离较远时。在其他方面一致的概率框架中,全基因组分析中的系统误差可追溯到模型错误设定问题,这表明应设计出更好的序列进化模型,即使在最具挑战性的条件下,该模型对树重建假象也应更具稳健性。
我们聚焦于后生动物树的先前系统发育基因组学研究中分析过的一个特征明确的LBA假象,在该研究中,两个快速进化的动物门,线虫和扁形动物,根据外类群的不同,要么出现在所有其他两侧对称动物的基部,要么出现在原口动物内部。我们将这个假象结果用作案例研究,以比较两种替代模型的稳健性:一种基于氨基酸替换经验矩阵(WAG)的标准位点均匀模型,以及一种位点异质混合模型(CAT)。同时,我们提出一种后验预测检验,用于衡量模型对序列饱和度的认知程度。
采用贝叶斯框架,我们表明当使用位点异质模型CAT时,在WAG下观察到的LBA假象消失。通过交叉验证,我们进一步证明在该数据集上CAT比WAG具有更好的统计拟合度。最后,使用我们的统计拟合优度检验,我们表明CAT(而非WAG)正确地考虑了总体饱和度水平,这是由于对位点特异性氨基酸偏好的更好估计。
CAT模型似乎比WAG模型对LBA假象更具稳健性,主要是因为它正确地预测了比对中每个位点氨基酸字母表有效大小较小所隐含的趋同和回复的高概率。更一般地说,我们的结果提供了有力证据,即替换过程中的位点特异性需要被考虑在内,以便获得更可靠的系统发育树。