Fogolari Federico, Pieri Lidia, Dovier Agostino, Bortolussi Luca, Giugliarelli Gilberto, Corazza Alessandra, Esposito Gennaro, Viglino Paolo
Dipartimento di Scienze e Tecnologie Biomediche, Università di Udine, Udine, Italy.
BMC Struct Biol. 2007 Mar 23;7:15. doi: 10.1186/1472-6807-7-15.
Reduced representations of proteins have been playing a keyrole in the study of protein folding. Many such models are available, with different representation detail. Although the usefulness of many such models for structural bioinformatics applications has been demonstrated in recent years, there are few intermediate resolution models endowed with an energy model capable, for instance, of detecting native or native-like structures among decoy sets. The aim of the present work is to provide a discrete empirical potential for a reduced protein model termed here PC2CA, because it employs a PseudoCovalent structure with only 2 Centers of interactions per Amino acid, suitable for protein model quality assessment.
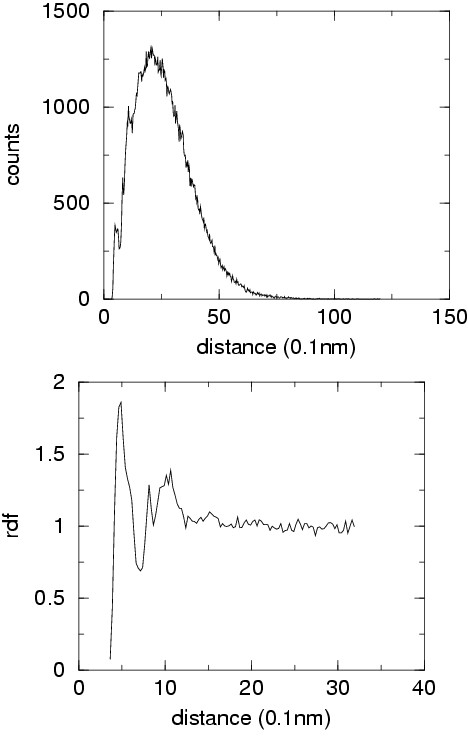
All protein structures in the set top500H have been converted in reduced form. The distribution of pseudobonds, pseudoangle, pseudodihedrals and distances between centers of interactions have been converted into potentials of mean force. A suitable reference distribution has been defined for non-bonded interactions which takes into account excluded volume effects and protein finite size. The correlation between adjacent main chain pseudodihedrals has been converted in an additional energetic term which is able to account for cooperative effects in secondary structure elements. Local energy surface exploration is performed in order to increase the robustness of the energy function.
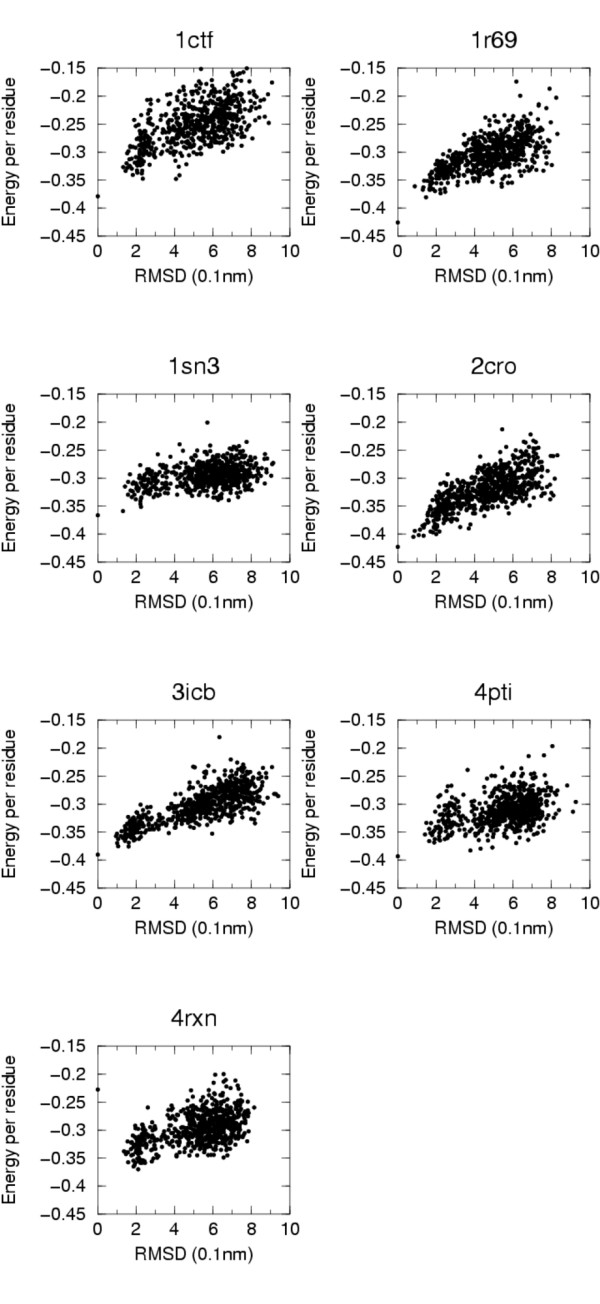
The model and the energy definition proposed have been tested on all the multiple decoys' sets in the Decoys'R'us database. The energetic model is able to recognize, for almost all sets, native-like structures (RMSD less than 2.0 A). These results and those obtained in the blind CASP7 quality assessment experiment suggest that the model compares well with scoring potentials with finer granularity and could be useful for fast exploration of conformational space. Parameters are available at the url: http://www.dstb.uniud.it/~ffogolari/download/.
简化的蛋白质表示在蛋白质折叠研究中一直发挥着关键作用。有许多这样的模型,其表示细节各不相同。尽管近年来已证明许多此类模型在结构生物信息学应用中的有用性,但很少有中间分辨率模型具备能量模型,例如能够在诱饵集中检测天然或类似天然的结构。本工作的目的是为一种名为PC2CA的简化蛋白质模型提供一种离散经验势,因为它采用了每个氨基酸仅具有2个相互作用中心的伪共价结构,适用于蛋白质模型质量评估。
top500H集中的所有蛋白质结构均已转换为简化形式。伪键、伪角、伪二面角以及相互作用中心之间的距离分布已转换为平均力势。已为非键相互作用定义了合适的参考分布,该分布考虑了排除体积效应和蛋白质有限大小。相邻主链伪二面角之间的相关性已转换为一个额外的能量项,该项能够解释二级结构元件中的协同效应。进行局部能量表面探索以提高能量函数的稳健性。
所提出的模型和能量定义已在Decoys'R'us数据库中的所有多个诱饵集上进行了测试。能量模型几乎能够识别所有集合中的类似天然结构(RMSD小于2.0埃)。这些结果以及在盲法CASP7质量评估实验中获得的结果表明,该模型与具有更精细粒度的评分势相比具有优势,并且可用于快速探索构象空间。参数可在以下网址获取:http://www.dstb.uniud.it/~ffogolari/download/ 。