Department of Bioinformatics, Institute of Biochemistry and Biophysics, University of Tehran, Tehran, Iran.
BMC Bioinformatics. 2010 Jan 9;11:16. doi: 10.1186/1471-2105-11-16.
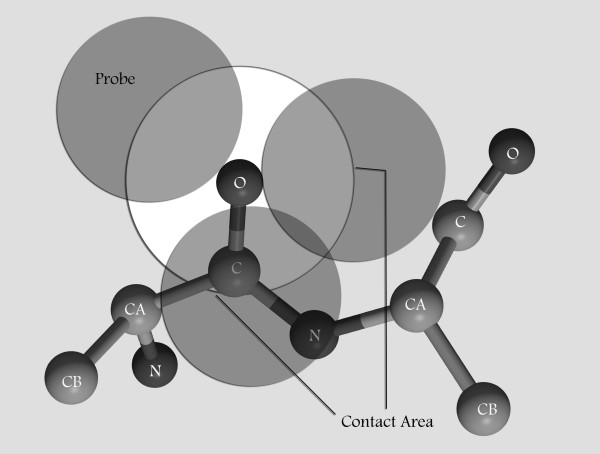
Considering energy function to detect a correct protein fold from incorrect ones is very important for protein structure prediction and protein folding. Knowledge-based mean force potentials are certainly the most popular type of interaction function for protein threading. They are derived from statistical analyses of interacting groups in experimentally determined protein structures. These potentials are developed at the atom or the amino acid level. Based on orientation dependent contact area, a new type of knowledge-based mean force potential has been developed.
We developed a new approach to calculate a knowledge-based potential of mean-force, using pairwise residue contact area. To test the performance of our approach, we performed it on several decoy sets to measure its ability to discriminate native structure from decoys. This potential has been able to distinguish native structures from the decoys in the most cases. Further, the calculated Z-scores were quite high for all protein datasets.
This knowledge-based potential of mean force can be used in protein structure prediction, fold recognition, comparative modelling and molecular recognition. The program is available at http://www.bioinf.cs.ipm.ac.ir/softwares/surfield.
考虑能量函数来从错误的结构中检测正确的蛋白质折叠对于蛋白质结构预测和蛋白质折叠非常重要。基于知识的平均力势是蛋白质穿线中最受欢迎的相互作用函数类型。它们是从实验确定的蛋白质结构中相互作用基团的统计分析中得出的。这些势是在原子或氨基酸水平上开发的。基于取向相关的接触面积,开发了一种新型基于知识的平均力势。
我们开发了一种新的方法来计算基于成对残基接触面积的基于知识的平均力势。为了测试我们方法的性能,我们在几个诱饵集上进行了测试,以衡量其区分天然结构与诱饵的能力。在大多数情况下,这种势能够将天然结构与诱饵区分开来。此外,对于所有蛋白质数据集,计算出的 Z 分数都相当高。
这种基于知识的平均力势可用于蛋白质结构预测、折叠识别、比较建模和分子识别。该程序可在 http://www.bioinf.cs.ipm.ac.ir/softwares/surfield 上获得。