Di Camillo Barbara, Toffolo Gianna, Nair Sreekumaran K, Greenlund Laura J, Cobelli Claudio
Information Engineering Department, University of Padova, 35131 Padova, Italy.
BMC Bioinformatics. 2007 Mar 8;8 Suppl 1(Suppl 1):S10. doi: 10.1186/1471-2105-8-S1-S10.
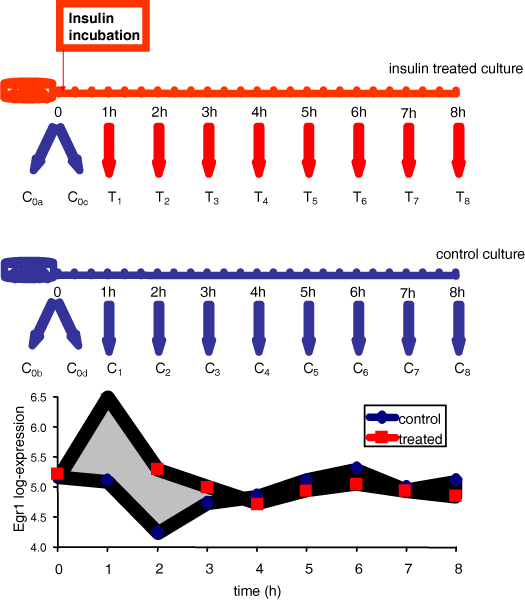
Microarray time series studies are essential to understand the dynamics of molecular events. In order to limit the analysis to those genes that change expression over time, a first necessary step is to select differentially expressed transcripts. A variety of methods have been proposed to this purpose; however, these methods are seldom applicable in practice since they require a large number of replicates, often available only for a limited number of samples. In this data-poor context, we evaluate the performance of three selection methods, using synthetic data, over a range of experimental conditions. Application to real data is also discussed.
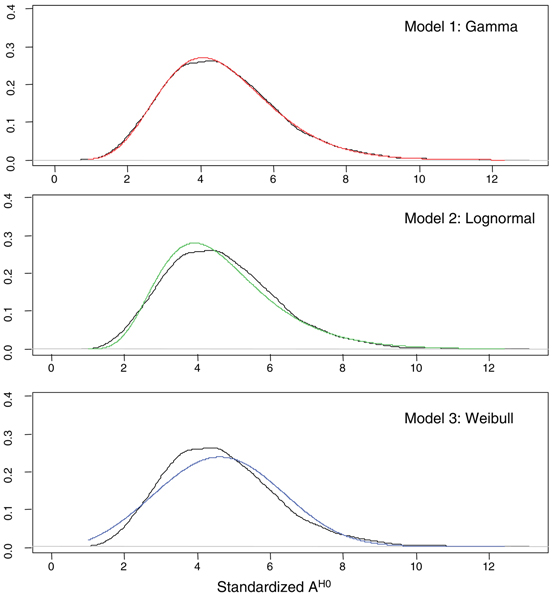
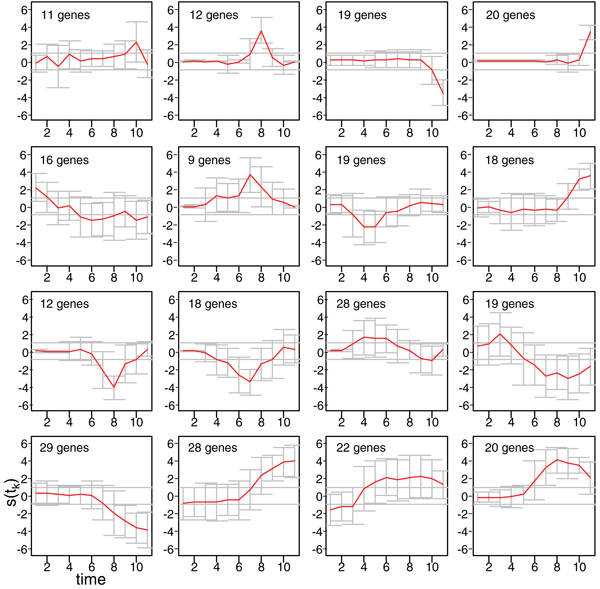
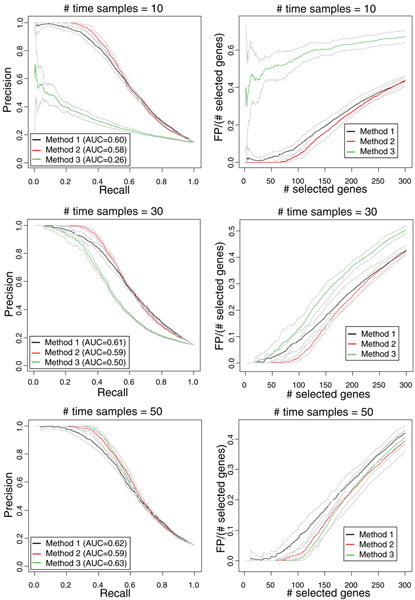
Three methods are considered, to assess differentially expressed genes in data-poor conditions. Method 1 uses a threshold on individual samples based on a model of the experimental error. Method 2 calculates the area of the region bounded by the time series expression profiles, and considers the gene differentially expressed if the area exceeds a threshold based on a model of the experimental error. These two methods are compared to Method 3, recently proposed in the literature, which exploits splines fit to compare time series profiles. Application of the three methods to synthetic data indicates that Method 2 outperforms the other two both in Precision and Recall when short time series are analyzed, while Method 3 outperforms the other two for long time series.
These results help to address the choice of the algorithm to be used in data-poor time series expression study, depending on the length of the time series.
微阵列时间序列研究对于理解分子事件的动态变化至关重要。为了将分析局限于那些随时间改变表达的基因,首要的必要步骤是选择差异表达的转录本。为此已经提出了多种方法;然而,这些方法在实际中很少适用,因为它们需要大量的重复样本,而这些样本通常仅适用于有限数量的样品。在这种数据匮乏的情况下,我们使用合成数据在一系列实验条件下评估了三种选择方法的性能。还讨论了这些方法在实际数据中的应用。
考虑了三种方法来评估数据匮乏条件下的差异表达基因。方法1基于实验误差模型对单个样本使用阈值。方法2计算由时间序列表达谱界定的区域面积,并根据实验误差模型,如果该面积超过阈值,则认为该基因差异表达。将这两种方法与文献中最近提出的方法3进行比较,方法3利用样条拟合来比较时间序列谱。将这三种方法应用于合成数据表明,在分析短时间序列时,方法2在精度和召回率方面均优于其他两种方法,而在分析长时间序列时,方法3优于其他两种方法。
这些结果有助于根据时间序列的长度,确定在数据匮乏的时间序列表达研究中使用的算法。