Kumar Charu G, Larson Joshua H, Band Mark R, Lewin Harris A
Laboratory of Mammalian Genome Biology, Department of Animal Sciences, University of Illinois at Urbana-Champaign, 210 Edward R. Madigan Laboratory, Urbana, IL 61801, USA.
BMC Genomics. 2007 May 9;8:113. doi: 10.1186/1471-2164-8-113.
Among the eutherian mammals, placental architecture varies to a greater extent than any other tissue. The diversity of placental types, even within a single mammalian order suggests that genes expressed in placenta are under strong Darwinian selection. Thus, the ruminant placenta may be a rich source of genes to explore adaptive evolutionary responses in mammals. The aim of our study was to identify novel transcripts expressed in ruminant placenta, and to characterize them with respect to their expression patterns, organization of coding sequences in the genome, and potential functions.
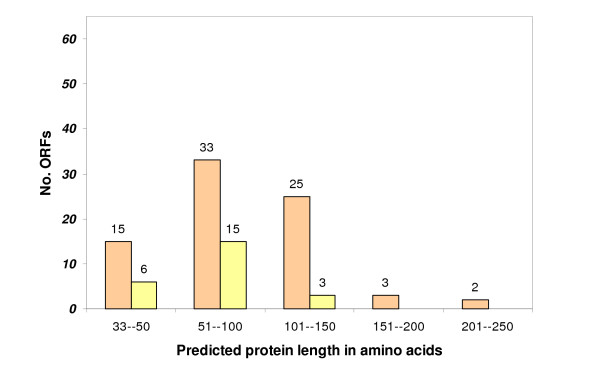
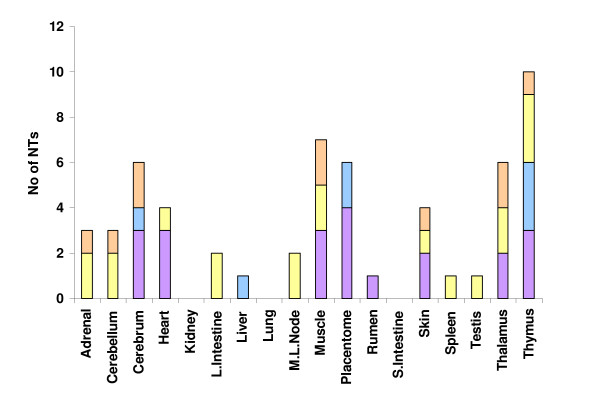
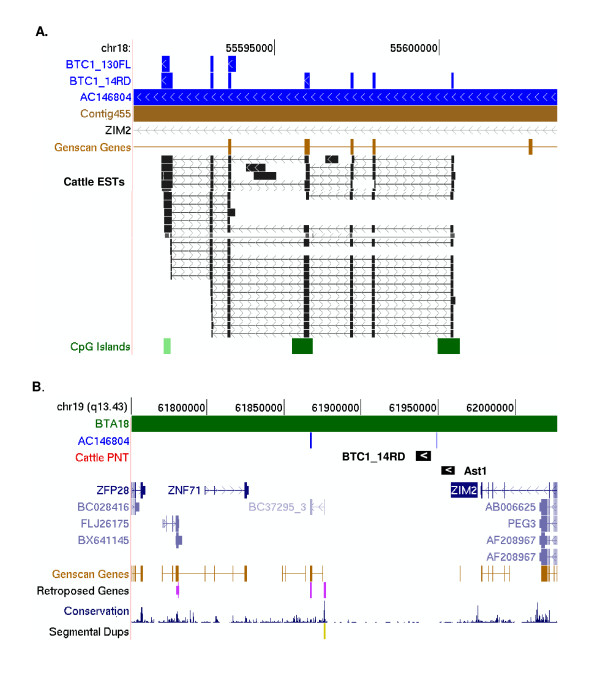
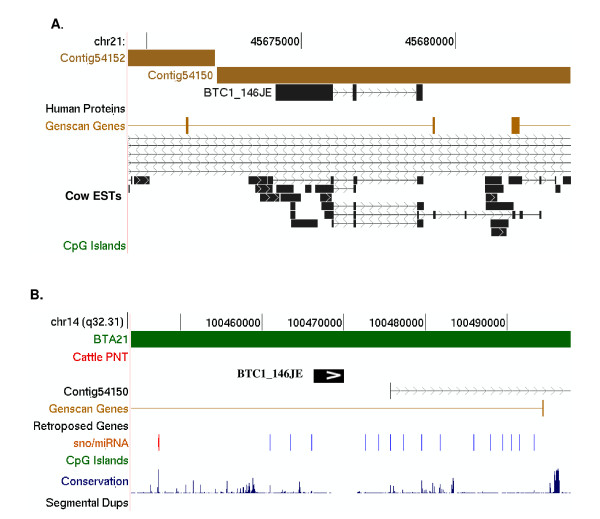
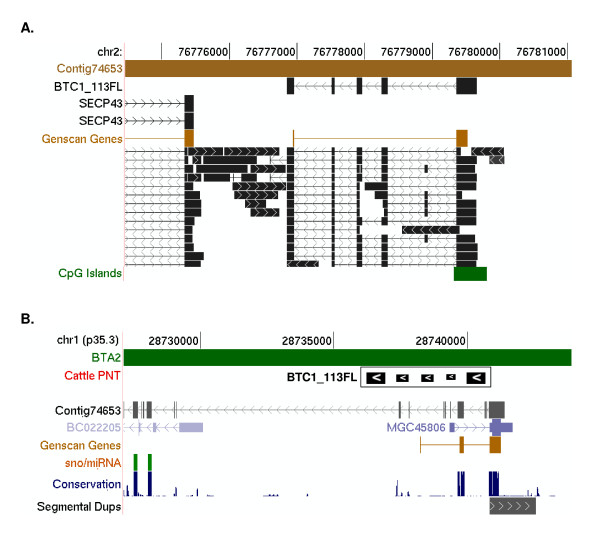
A combination of bioinformatics, comparative genomics and transcript profiling was used to identify and characterize 91 novel transcripts (NTs) represented in a cattle placenta cDNA library. These NTs have no significant similarity to any non-ferungulate DNA or RNA sequence. Proteins longer than 100 aa were predicted for 29 NTs, and 21 are candidate non-coding RNAs. Eighty-six NTs were found to be expressed in one or more of 18 different tissues, with 39 (42%) showing tissue-preference, including six that were expressed exclusively in placentome. The authenticity of the NTs was confirmed by their alignment to cattle genome sequence, 42 of which showed evidence of mRNA splicing. Analysis of the genomic context where NT genes reside revealed 61 to be in intergenic regions, whereas 30 are within introns of known genes. The genes encoding the NTs were found to be significantly associated with subtelomeric regions.
The 91 lineage-specific transcripts are a useful resource for studying adaptive evolutionary responses of the ruminant placenta. The presence of so many genes encoding NTs in cattle but not primates or rodents suggests that gene loss and gain are important mechanisms of genome evolution in mammals. Furthermore, the clustering of NT genes within subtelomeric regions suggests that such regions are highly dynamic and may foster the birth of novel genes. The sequencing of additional vertebrate genomes with defined phylogenetic relationships will permit the search for lineage-specific genes to take on a more evolutionary context that is required to understand their origins and functions.
在真兽类哺乳动物中,胎盘结构的差异程度比其他任何组织都要大。胎盘类型的多样性,即使在单一哺乳动物目内也是如此,这表明在胎盘中表达的基因受到强烈的达尔文选择。因此,反刍动物胎盘可能是探索哺乳动物适应性进化反应的丰富基因来源。我们研究的目的是鉴定反刍动物胎盘中表达的新转录本,并根据它们的表达模式、基因组中编码序列的组织方式以及潜在功能对其进行表征。
结合生物信息学、比较基因组学和转录谱分析,鉴定并表征了牛胎盘cDNA文库中代表的91个新转录本(NTs)。这些NTs与任何非有蹄类动物的DNA或RNA序列均无显著相似性。预测有29个NTs编码的蛋白质长度超过100个氨基酸,21个是候选非编码RNA。发现86个NTs在18种不同组织中的一种或多种中表达,其中39个(42%)表现出组织偏好性,包括6个仅在胎盘小叶中表达的。通过与牛基因组序列比对证实了NTs的真实性,其中42个显示出mRNA剪接的证据。对NTs基因所在的基因组背景分析表明,61个位于基因间区域,而30个位于已知基因的内含子内。发现编码NTs的基因与亚端粒区域显著相关。
这91个谱系特异性转录本是研究反刍动物胎盘适应性进化反应的有用资源。牛中存在如此多编码NTs的基因,而灵长类动物或啮齿动物中却没有,这表明基因的丢失和获得是哺乳动物基因组进化的重要机制。此外,NTs基因在亚端粒区域的聚集表明这些区域高度动态,可能促进新基因的产生。对具有明确系统发育关系的其他脊椎动物基因组进行测序,将使寻找谱系特异性基因的研究具有更进化的背景,这对于理解它们的起源和功能是必需的。