Flores Samuel C, Lu Long J, Yang Julie, Carriero Nicholas, Gerstein Mark B
Department of Physics, Yale University, New Haven, CT, USA.
BMC Bioinformatics. 2007 May 22;8:167. doi: 10.1186/1471-2105-8-167.
Relating features of protein sequences to structural hinges is important for identifying domain boundaries, understanding structure-function relationships, and designing flexibility into proteins. Efforts in this field have been hampered by the lack of a proper dataset for studying characteristics of hinges.
Using the Molecular Motions Database we have created a Hinge Atlas of manually annotated hinges and a statistical formalism for calculating the enrichment of various types of residues in these hinges.
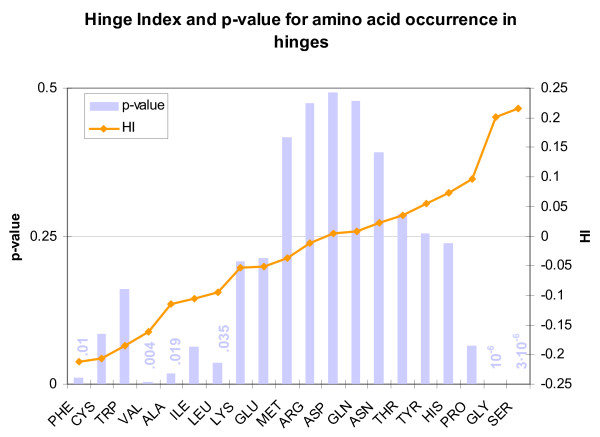
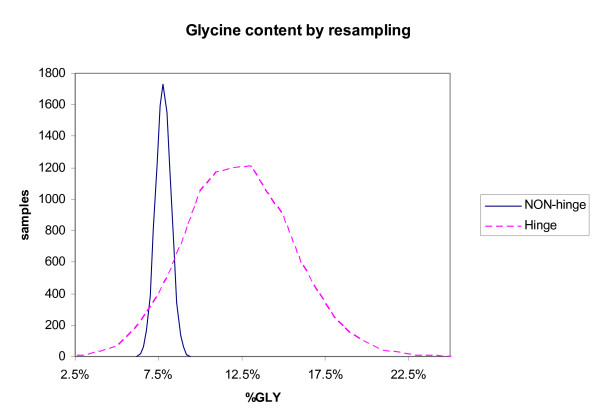
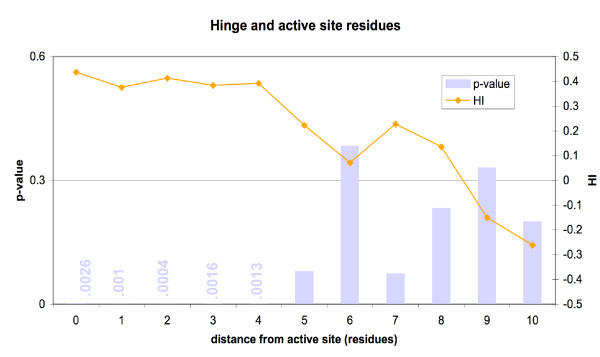
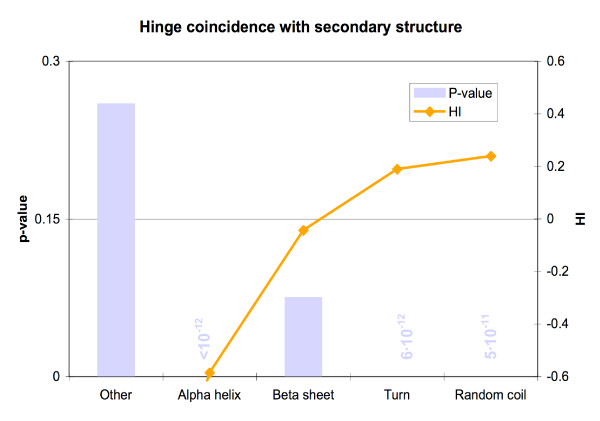
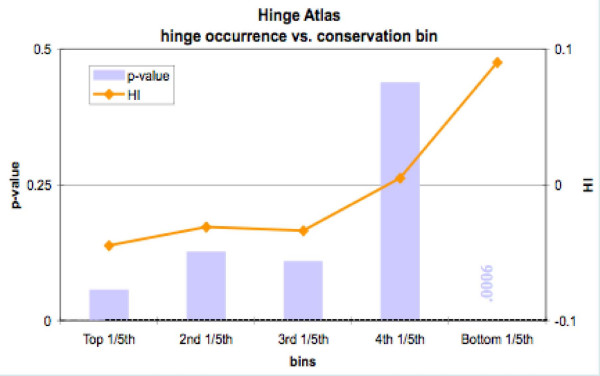
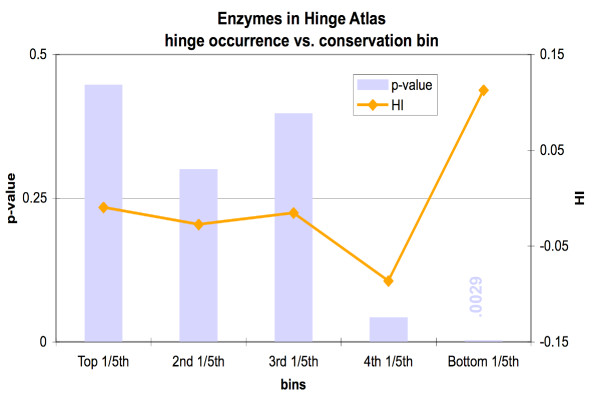
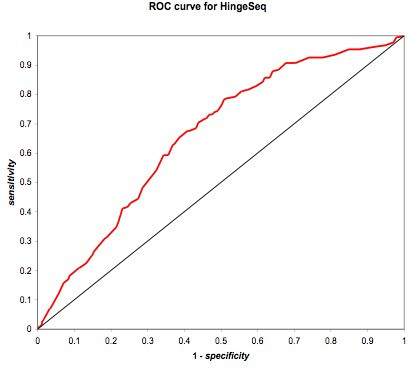
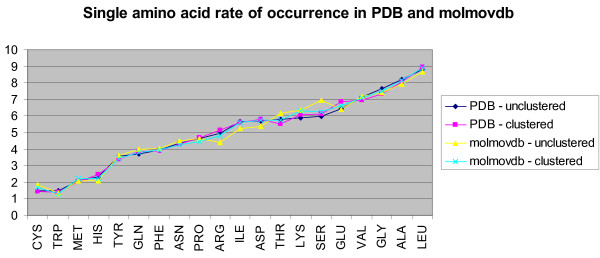
We found various correlations between hinges and sequence features. Some of these are expected; for instance, we found that hinges tend to occur on the surface and in coils and turns and to be enriched with small and hydrophilic residues. Others are less obvious and intuitive. In particular, we found that hinges tend to coincide with active sites, but unlike the latter they are not at all conserved in evolution. We evaluate the potential for hinge prediction based on sequence. Motions play an important role in catalysis and protein-ligand interactions. Hinge bending motions comprise the largest class of known motions. Therefore it is important to relate the hinge location to sequence features such as residue type, physicochemical class, secondary structure, solvent exposure, evolutionary conservation, and proximity to active sites. To do this, we first generated the Hinge Atlas, a set of protein motions with the hinge locations manually annotated, and then studied the coincidence of these features with the hinge location. We found that all of the features have bearing on the hinge location. Most interestingly, we found that hinges tend to occur at or near active sites and yet unlike the latter are not conserved. Less surprisingly, we found that hinge residues tend to be small, not hydrophobic or aliphatic, and occur in turns and random coils on the surface. A functional sequence based hinge predictor was made which uses some of the data generated in this study. The Hinge Atlas is made available to the community for further flexibility studies.
将蛋白质序列特征与结构铰链相关联,对于识别结构域边界、理解结构 - 功能关系以及在蛋白质中设计灵活性而言至关重要。该领域的研究工作因缺乏用于研究铰链特征的合适数据集而受阻。
利用分子运动数据库,我们创建了一个手动注释铰链的铰链图谱以及一种统计形式,用于计算这些铰链中各类残基的富集情况。
我们发现了铰链与序列特征之间的各种相关性。其中一些是预期的;例如,我们发现铰链倾向于出现在表面、卷曲和转角处,并且富含小的亲水性残基。其他的则不那么明显和直观。特别是,我们发现铰链倾向于与活性位点重合,但与后者不同的是,它们在进化过程中一点也不保守。我们评估了基于序列进行铰链预测的潜力。运动在催化和蛋白质 - 配体相互作用中起着重要作用。铰链弯曲运动是已知运动中最大的一类。因此,将铰链位置与诸如残基类型、物理化学类别、二级结构、溶剂暴露、进化保守性以及与活性位点的接近程度等序列特征相关联是很重要的。为此,我们首先生成了铰链图谱,这是一组手动注释了铰链位置的蛋白质运动,然后研究了这些特征与铰链位置的重合情况。我们发现所有这些特征都与铰链位置有关。最有趣的是,我们发现铰链倾向于出现在活性位点或其附近,但与后者不同的是并不保守。不太令人惊讶的是,我们发现铰链残基往往较小,不是疏水性或脂肪族的,并且出现在表面的转角和无规卷曲中。基于本研究生成的一些数据制作了一个基于功能序列的铰链预测器。铰链图谱已提供给科学界用于进一步的灵活性研究。