Moriya Yuki, Itoh Masumi, Okuda Shujiro, Yoshizawa Akiyasu C, Kanehisa Minoru
Bioinformatics Center, Institute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan.
Nucleic Acids Res. 2007 Jul;35(Web Server issue):W182-5. doi: 10.1093/nar/gkm321. Epub 2007 May 25.
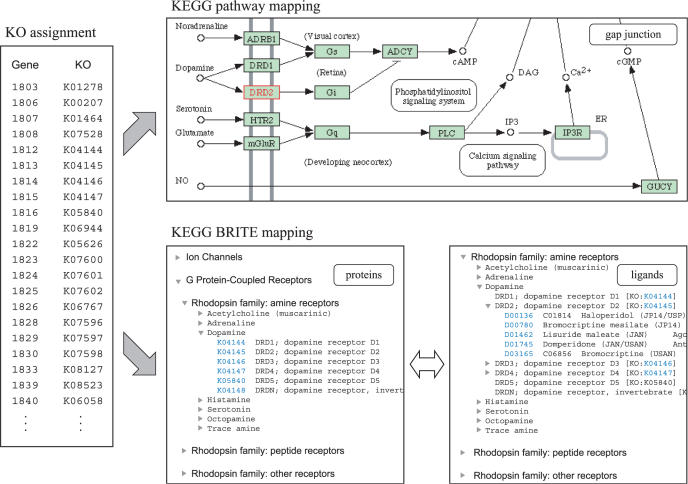
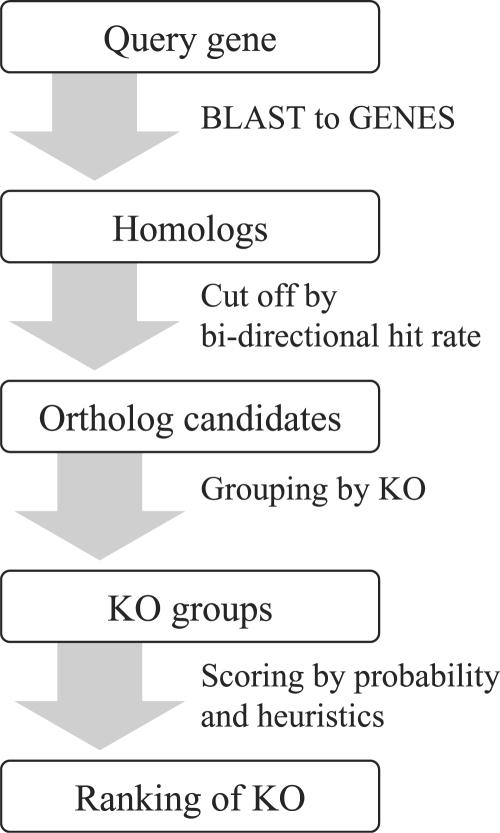
The number of complete and draft genomes is rapidly growing in recent years, and it has become increasingly important to automate the identification of functional properties and biological roles of genes in these genomes. In the KEGG database, genes in complete genomes are annotated with the KEGG orthology (KO) identifiers, or the K numbers, based on the best hit information using Smith-Waterman scores as well as by the manual curation. Each K number represents an ortholog group of genes, and it is directly linked to an object in the KEGG pathway map or the BRITE functional hierarchy. Here, we have developed a web-based server called KAAS (KEGG Automatic Annotation Server: http://www.genome.jp/kegg/kaas/) i.e. an implementation of a rapid method to automatically assign K numbers to genes in the genome, enabling reconstruction of KEGG pathways and BRITE hierarchies. The method is based on sequence similarities, bi-directional best hit information and some heuristics, and has achieved a high degree of accuracy when compared with the manually curated KEGG GENES database.
近年来,完整基因组和草图基因组的数量在迅速增长,因此,对这些基因组中基因的功能特性和生物学作用进行自动识别变得越来越重要。在KEGG数据库中,基于使用Smith-Waterman评分的最佳匹配信息以及人工整理,完整基因组中的基因用KEGG直系同源物(KO)标识符或K编号进行注释。每个K编号代表一个基因直系同源物组,并且它与KEGG通路图或BRITE功能层次结构中的一个对象直接相关。在这里,我们开发了一个基于网络的服务器,称为KAAS(KEGG自动注释服务器:http://www.genome.jp/kegg/kaas/),即一种快速方法的实现,该方法可自动为基因组中的基因分配K编号,从而能够重建KEGG通路和BRITE层次结构。该方法基于序列相似性、双向最佳匹配信息和一些启发式方法,与人工整理的KEGG GENES数据库相比,具有很高的准确性。