Hsieh Wen-Ping, Passador-Gurgel Gisele, Stone Eric A, Gibson Greg
Department of Genetics, Gardner Hall, North Carolina State University, Raleigh, North Carolina 27695-7614, USA.
Genome Biol. 2007;8(6):R98. doi: 10.1186/gb-2007-8-6-r98.
Populations diverge in genotype and phenotype under the influence of such evolutionary processes as genetic drift, mutation accumulation, and natural selection. Because genotype maps onto phenotype by way of transcription, it is of interest to evaluate how these evolutionary factors influence the structure of variation at the level of transcription. Here, we explore the distributions of cis-acting and trans-acting factors and their relative contributions to expression of transcripts that exhibit two or more classes of abundance among individuals within populations.
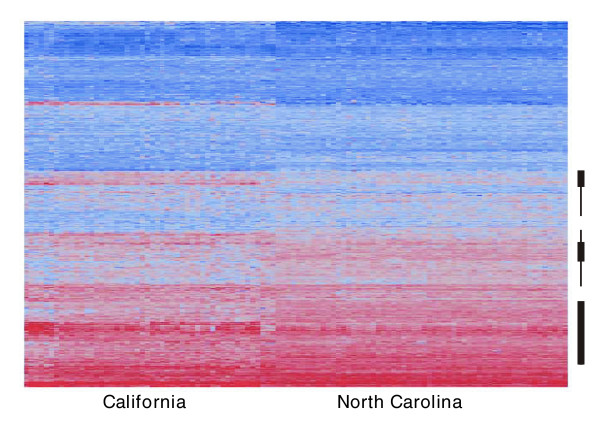
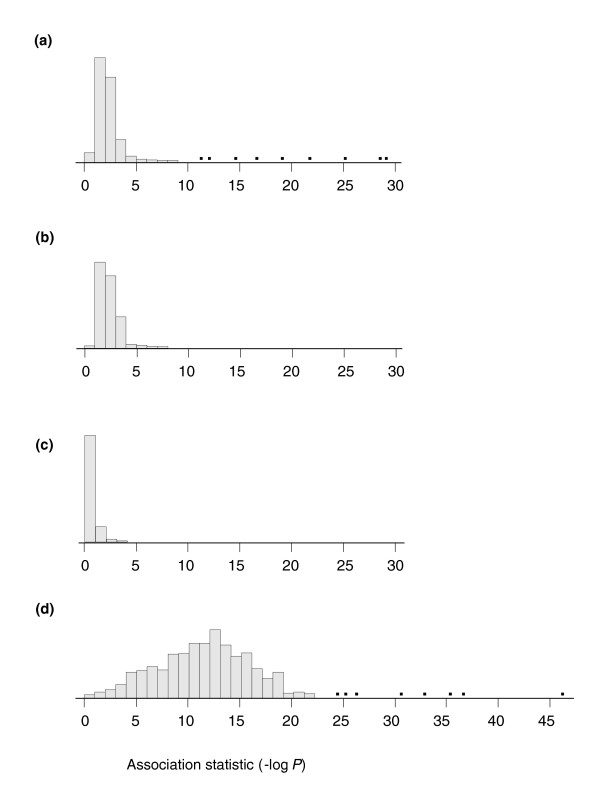
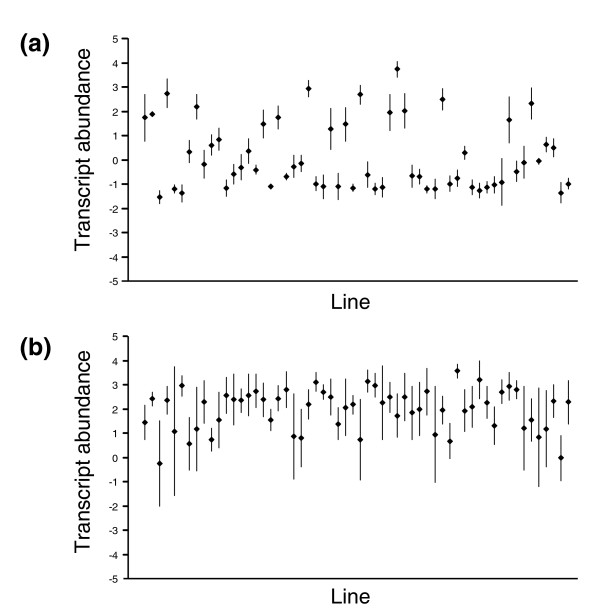
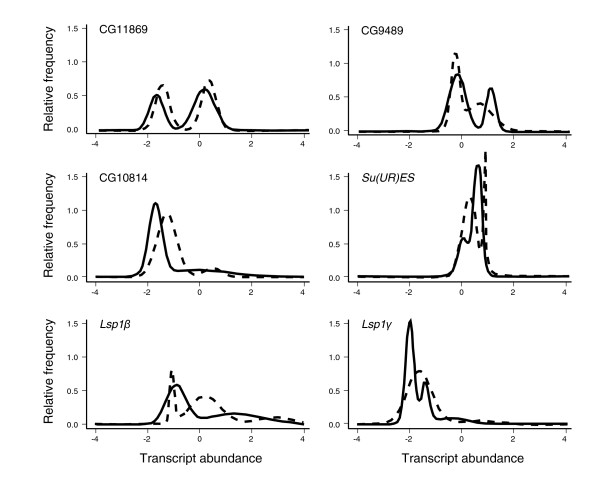
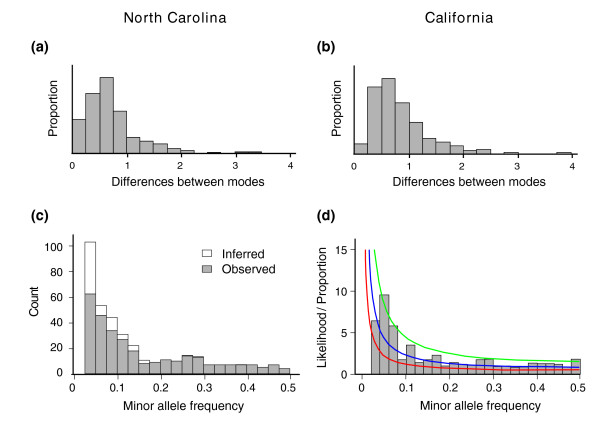
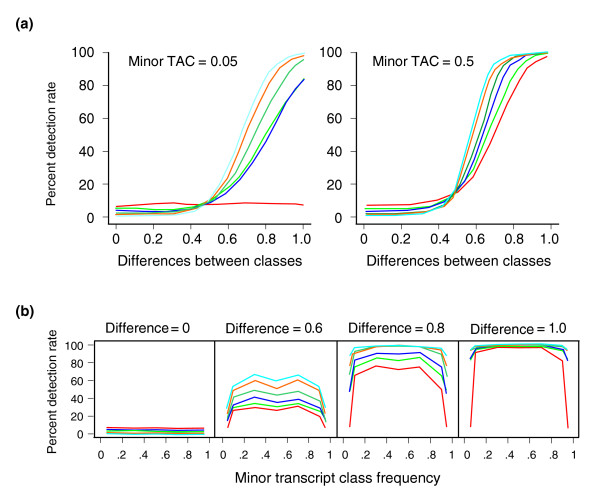
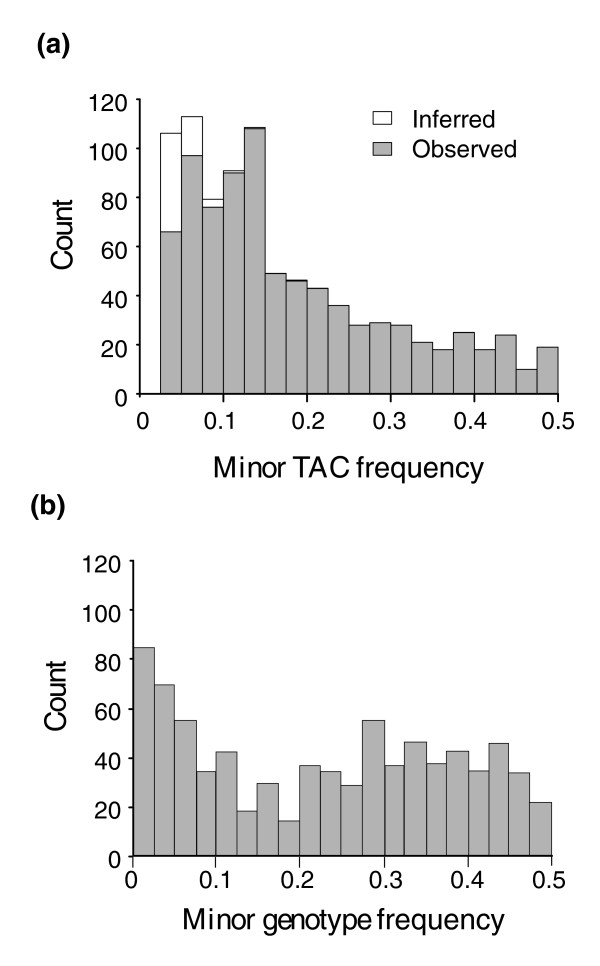
Expression profiling using cDNA microarrays was conducted in Drosophila melanogaster adult female heads for 58 nearly isogenic lines from a North Carolina population and 50 from a California population. Using a mixture modeling approach, transcripts were identified that exhibit more than one mode of transcript abundance across the samples. Power studies indicate that sample sizes of 50 individuals will generally be sufficient to detect divergent transcript abundance classes. The distribution of transcript abundance classes is skewed toward low frequency minor classes, which is reminiscent of the typical skew in genotype frequencies. Similar results are observed in reported data on gene expression in human lymphoblast cell lines, in which analysis of association with linked polymorphisms implies that cis-acting single nucleotide polymorphisms make only a modest contribution to bimodal distributions of transcript abundance.
Population surveys of gene expression may complement genetical genomics as a general approach to quantifying sources of transcriptional variation. Differential expression of transcripts among individuals is due to a complex interplay of cis-acting and trans-acting factors.
在遗传漂变、突变积累和自然选择等进化过程的影响下,种群在基因型和表型上会出现分化。由于基因型通过转录作用于表型,因此评估这些进化因素如何影响转录水平的变异结构就显得很有意义。在这里,我们探讨了顺式作用和反式作用因子的分布及其对种群内个体间呈现两类或更多丰度类别的转录本表达的相对贡献。
利用cDNA微阵列对来自北卡罗来纳种群的58个近等基因系和来自加利福尼亚种群的50个近等基因系的黑腹果蝇成年雌性头部进行了表达谱分析。使用混合建模方法,鉴定出在所有样本中呈现不止一种转录本丰度模式的转录本。功效研究表明,50个个体的样本量通常足以检测到不同的转录本丰度类别。转录本丰度类别的分布偏向低频的次要类别,这让人联想到基因型频率的典型偏态。在关于人类淋巴母细胞系基因表达的报道数据中也观察到了类似结果,其中与连锁多态性的关联分析表明,顺式作用单核苷酸多态性对转录本丰度的双峰分布仅起适度作用。
基因表达的种群调查作为一种量化转录变异来源的通用方法,可能会补充遗传基因组学。个体间转录本的差异表达是顺式作用和反式作用因子复杂相互作用的结果。