Stranger Barbara E, Forrest Matthew S, Clark Andrew G, Minichiello Mark J, Deutsch Samuel, Lyle Robert, Hunt Sarah, Kahl Brenda, Antonarakis Stylianos E, Tavaré Simon, Deloukas Panagiotis, Dermitzakis Emmanouil T
Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, United Kingdom.
PLoS Genet. 2005 Dec;1(6):e78. doi: 10.1371/journal.pgen.0010078. Epub 2005 Dec 16.
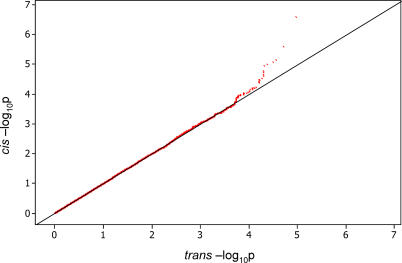
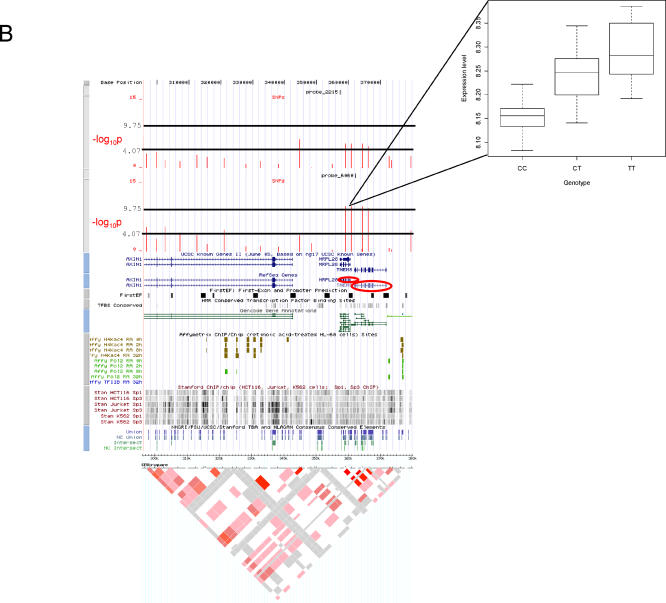
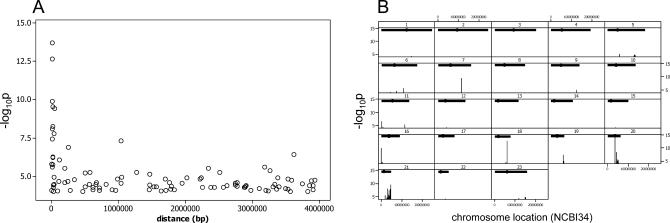
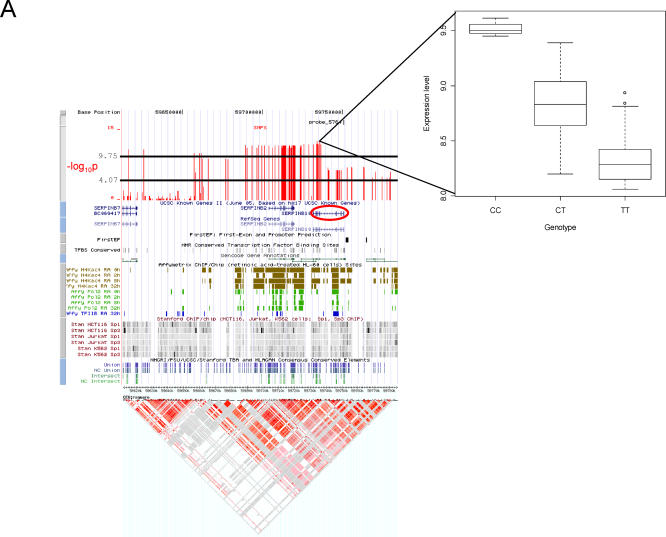
The exploration of quantitative variation in human populations has become one of the major priorities for medical genetics. The successful identification of variants that contribute to complex traits is highly dependent on reliable assays and genetic maps. We have performed a genome-wide quantitative trait analysis of 630 genes in 60 unrelated Utah residents with ancestry from Northern and Western Europe using the publicly available phase I data of the International HapMap project. The genes are located in regions of the human genome with elevated functional annotation and disease interest including the ENCODE regions spanning 1% of the genome, Chromosome 21 and Chromosome 20q12-13.2. We apply three different methods of multiple test correction, including Bonferroni, false discovery rate, and permutations. For the 374 expressed genes, we find many regions with statistically significant association of single nucleotide polymorphisms (SNPs) with expression variation in lymphoblastoid cell lines after correcting for multiple tests. Based on our analyses, the signal proximal (cis-) to the genes of interest is more abundant and more stable than distal and trans across statistical methodologies. Our results suggest that regulatory polymorphism is widespread in the human genome and show that the 5-kb (phase I) HapMap has sufficient density to enable linkage disequilibrium mapping in humans. Such studies will significantly enhance our ability to annotate the non-coding part of the genome and interpret functional variation. In addition, we demonstrate that the HapMap cell lines themselves may serve as a useful resource for quantitative measurements at the cellular level.
对人类群体中数量变异的探索已成为医学遗传学的主要优先事项之一。成功鉴定出影响复杂性状的变异高度依赖于可靠的检测方法和遗传图谱。我们利用国际人类基因组单体型图计划(International HapMap project)公开的一期数据,对60名具有北欧和西欧血统的犹他州无关居民中的630个基因进行了全基因组数量性状分析。这些基因位于人类基因组中功能注释丰富且与疾病相关的区域,包括占基因组1%的ENCODE区域、21号染色体以及20号染色体的q12 - 13.2区域。我们应用了三种不同的多重检验校正方法,包括邦费罗尼校正、错误发现率校正和置换检验。对于374个表达基因,在进行多重检验校正后,我们发现许多区域的单核苷酸多态性(SNP)与淋巴母细胞系中的表达变异存在统计学显著关联。基于我们的分析,在各种统计方法中,与感兴趣基因近端(顺式)的信号比远端和反式的信号更丰富、更稳定。我们的结果表明调控多态性在人类基因组中广泛存在,并表明5千碱基对(一期)的单体型图具有足够的密度,能够在人类中进行连锁不平衡作图。此类研究将显著增强我们注释基因组非编码部分并解释功能变异的能力。此外,我们证明单体型图细胞系本身可作为细胞水平定量测量的有用资源。