Toedling Joern, Skylar Oleg, Krueger Tammo, Fischer Jenny J, Sperling Silke, Huber Wolfgang
EMBL European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK.
BMC Bioinformatics. 2007 Jun 26;8:221. doi: 10.1186/1471-2105-8-221.
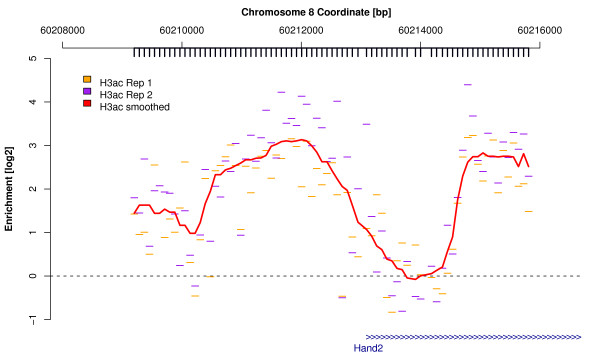
Chromatin immunoprecipitation combined with DNA microarrays (ChIP-chip) is a high-throughput assay for DNA-protein-binding or post-translational chromatin/histone modifications. However, the raw microarray intensity readings themselves are not immediately useful to researchers, but require a number of bioinformatic analysis steps. Identified enriched regions need to be bioinformatically annotated and compared to related datasets by statistical methods.
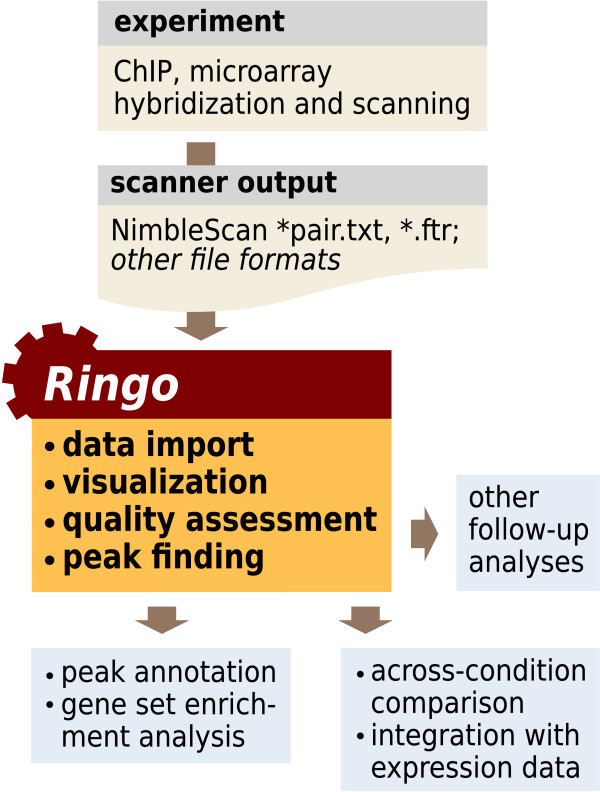
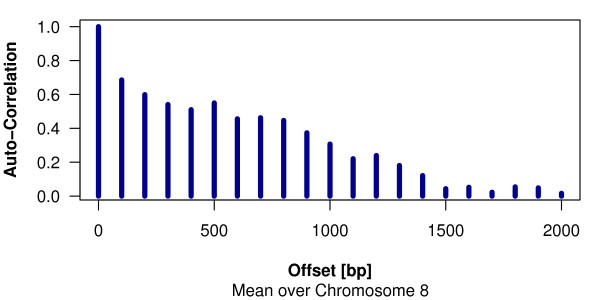
We present a free, open-source R package Ringo that facilitates the analysis of ChIP-chip experiments by providing functionality for data import, quality assessment, normalization and visualization of the data, and the detection of ChIP-enriched genomic regions.
Ringo integrates with other packages of the Bioconductor project, uses common data structures and is accompanied by ample documentation. It facilitates the construction of programmed analysis workflows, offers benefits in scalability, reproducibility and methodical scope of the analyses and opens up a broad selection of follow-up statistical and bioinformatic methods.
染色质免疫沉淀结合DNA微阵列技术(ChIP-chip)是一种用于检测DNA-蛋白质结合或翻译后染色质/组蛋白修饰的高通量分析方法。然而,原始的微阵列强度读数本身对研究人员并无直接用处,而是需要一系列生物信息学分析步骤。识别出的富集区域需要进行生物信息学注释,并通过统计方法与相关数据集进行比较。
我们提供了一个免费的开源R包Ringo,它通过提供数据导入、质量评估、数据标准化和可视化以及检测ChIP富集基因组区域的功能,促进了ChIP-chip实验的分析。
Ringo与Bioconductor项目的其他包集成,使用常见的数据结构,并附有丰富的文档。它有助于构建程序化的分析工作流程,在分析的可扩展性、可重复性和方法范围方面具有优势,并提供了广泛的后续统计和生物信息学方法选择。