Daraselia Nikolai, Yuryev Anton, Egorov Sergei, Mazo Ilya, Ispolatov Iaroslav
Ariadne Genomics, Inc, Rockville, MD 20850, USA.
BMC Bioinformatics. 2007 Jul 10;8:243. doi: 10.1186/1471-2105-8-243.
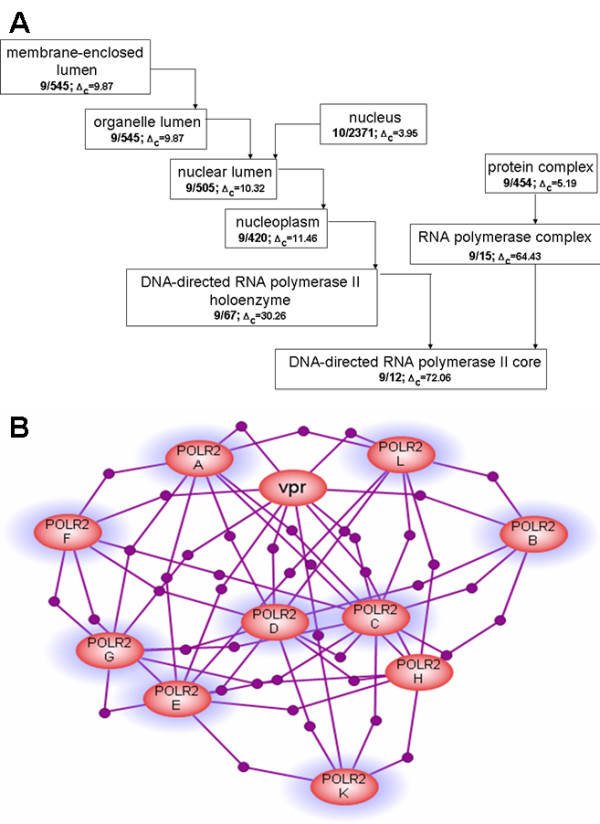
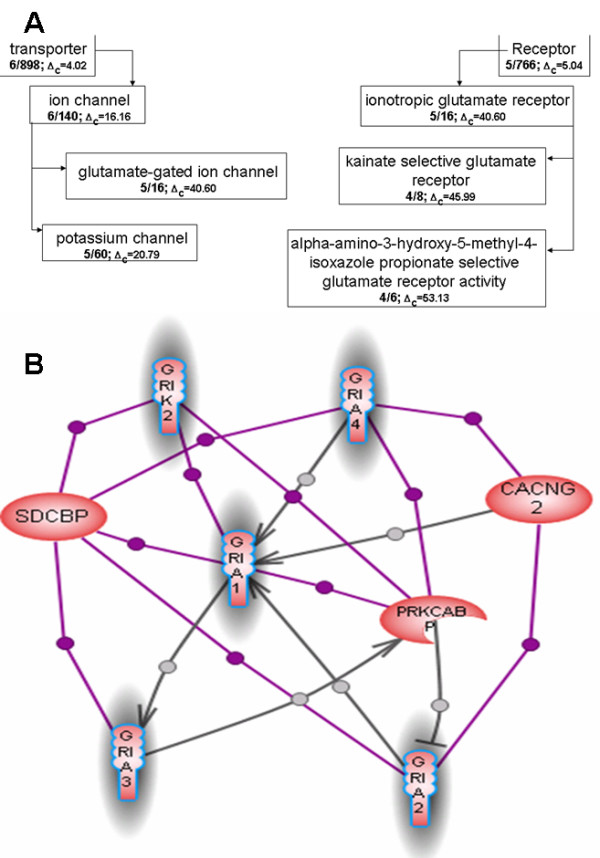
Uncovering cellular roles of a protein is a task of tremendous importance and complexity that requires dedicated experimental work as well as often sophisticated data mining and processing tools. Protein functions, often referred to as its annotations, are believed to manifest themselves through topology of the networks of inter-proteins interactions. In particular, there is a growing body of evidence that proteins performing the same function are more likely to interact with each other than with proteins with other functions. However, since functional annotation and protein network topology are often studied separately, the direct relationship between them has not been comprehensively demonstrated. In addition to having the general biological significance, such demonstration would further validate the data extraction and processing methods used to compose protein annotation and protein-protein interactions datasets.
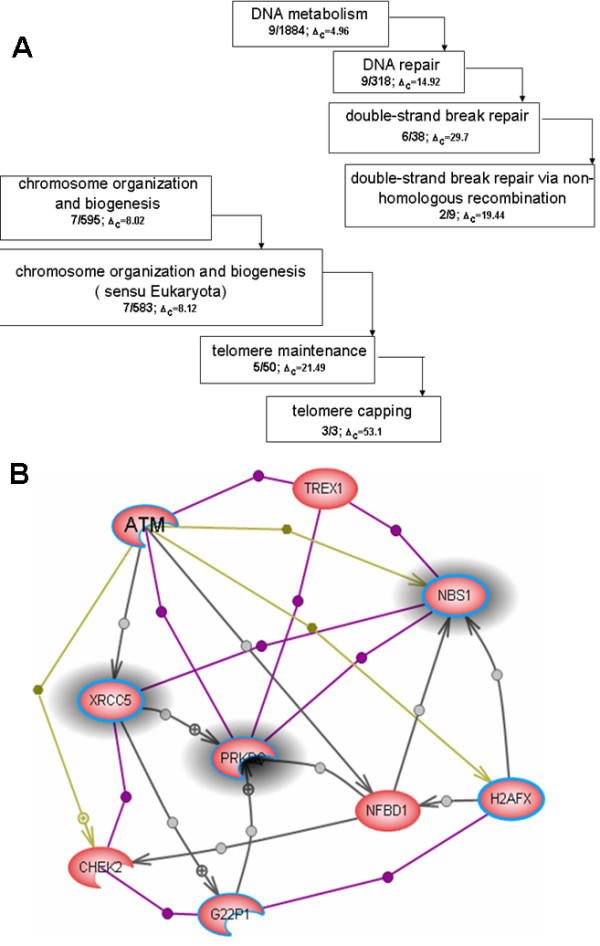
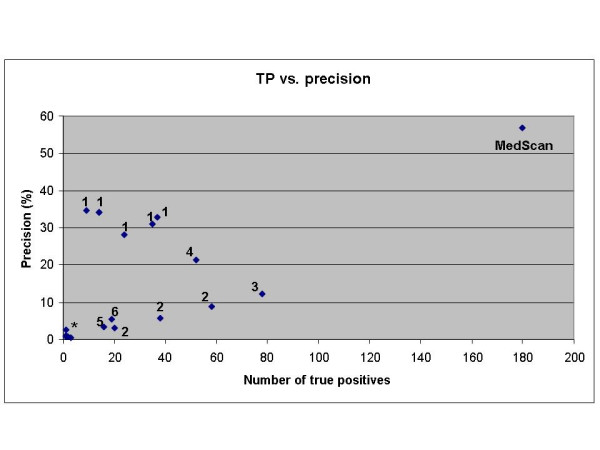
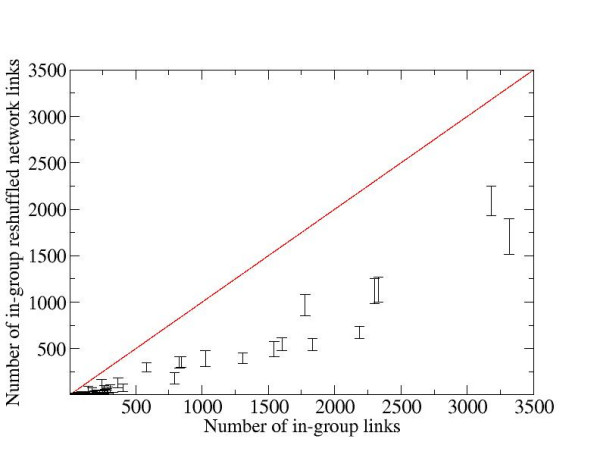
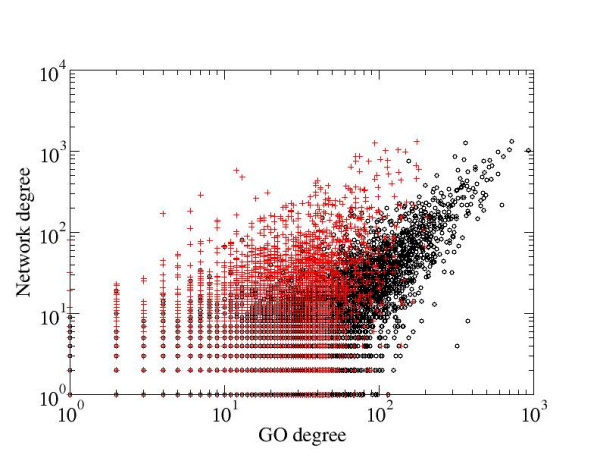
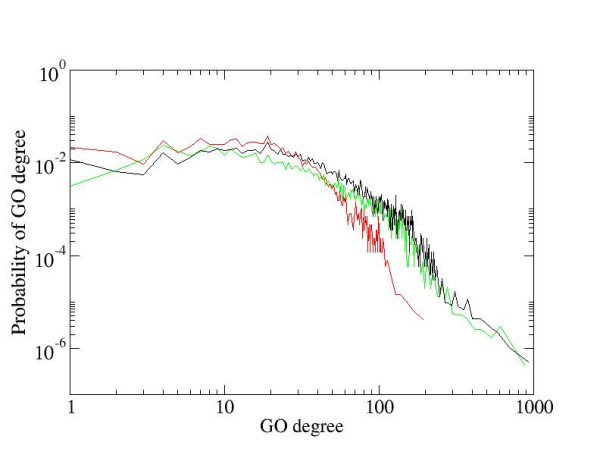
We developed a method for automatic extraction of protein functional annotation from scientific text based on the Natural Language Processing (NLP) technology. For the protein annotation extracted from the entire PubMed, we evaluated the precision and recall rates, and compared the performance of the automatic extraction technology to that of manual curation used in public Gene Ontology (GO) annotation. In the second part of our presentation, we reported a large-scale investigation into the correspondence between communities in the literature-based protein networks and GO annotation groups of functionally related proteins. We found a comprehensive two-way match: proteins within biological annotation groups form significantly denser linked network clusters than expected by chance and, conversely, densely linked network communities exhibit a pronounced non-random overlap with GO groups. We also expanded the publicly available GO biological process annotation using the relations extracted by our NLP technology. An increase in the number and size of GO groups without any noticeable decrease of the link density within the groups indicated that this expansion significantly broadens the public GO annotation without diluting its quality. We revealed that functional GO annotation correlates mostly with clustering in a physical interaction protein network, while its overlap with indirect regulatory network communities is two to three times smaller.
Protein functional annotations extracted by the NLP technology expand and enrich the existing GO annotation system. The GO functional modularity correlates mostly with the clustering in the physical interaction network, suggesting that the essential role of structural organization maintained by these interactions. Reciprocally, clustering of proteins in physical interaction networks can serve as an evidence for their functional similarity.
揭示一种蛋白质的细胞功能是一项极为重要且复杂的任务,需要专门的实验工作以及通常复杂的数据挖掘和处理工具。蛋白质功能,通常被称为其注释,被认为是通过蛋白质间相互作用网络的拓扑结构来体现的。特别是,越来越多的证据表明,执行相同功能的蛋白质比执行其他功能的蛋白质更有可能相互作用。然而,由于功能注释和蛋白质网络拓扑结构通常是分开研究的,它们之间的直接关系尚未得到全面证明。除了具有一般生物学意义外,这种证明还将进一步验证用于构建蛋白质注释和蛋白质 - 蛋白质相互作用数据集的数据提取和处理方法。
我们基于自然语言处理(NLP)技术开发了一种从科学文本中自动提取蛋白质功能注释的方法。对于从整个PubMed中提取的蛋白质注释,我们评估了精确率和召回率,并将自动提取技术的性能与公共基因本体(GO)注释中使用的人工整理性能进行了比较。在我们展示的第二部分,我们报告了对基于文献的蛋白质网络中的群落与功能相关蛋白质的GO注释组之间对应关系的大规模调查。我们发现了全面的双向匹配:生物学注释组内的蛋白质形成的连接网络簇比随机预期的要密集得多,相反,紧密连接的网络群落与GO组表现出明显的非随机重叠。我们还使用我们的NLP技术提取的关系扩展了公开可用的GO生物学过程注释。GO组数量和大小的增加而组内连接密度没有任何明显下降表明,这种扩展显著拓宽了公共GO注释而没有稀释其质量。我们发现GO功能注释主要与物理相互作用蛋白质网络中的聚类相关,而其与间接调控网络群落的重叠要小三分之二。
通过NLP技术提取的蛋白质功能注释扩展并丰富了现有的GO注释系统。GO功能模块性主要与物理相互作用网络中的聚类相关,表明这些相互作用维持的结构组织起着至关重要的作用。相反,蛋白质在物理相互作用网络中的聚类可以作为它们功能相似性的证据。