Institute of Biomedical Chemistry, RAMS, Moscow, Russia ; Ariadne Diagnostics LLC, Rockville, Maryland, United States of America.
Ariadne Diagnostics LLC, Rockville, Maryland, United States of America.
PLoS One. 2014 Jan 9;9(1):e84955. doi: 10.1371/journal.pone.0084955. eCollection 2014.
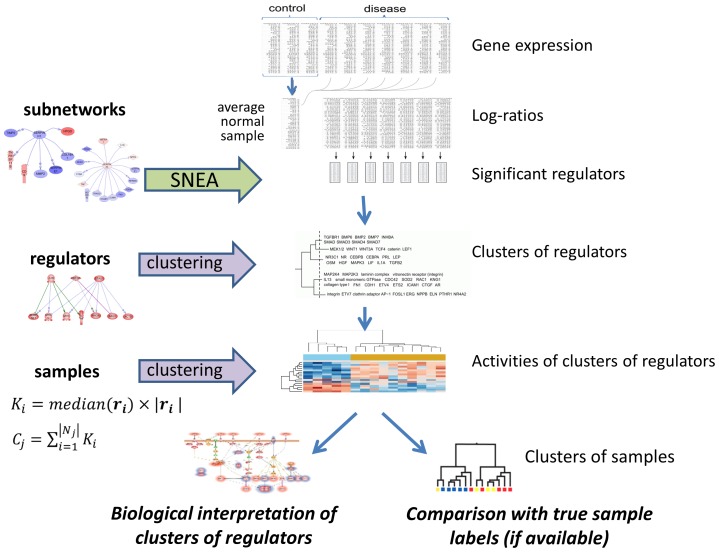
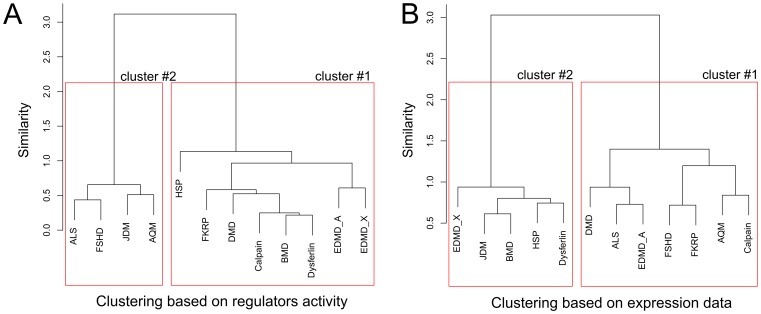
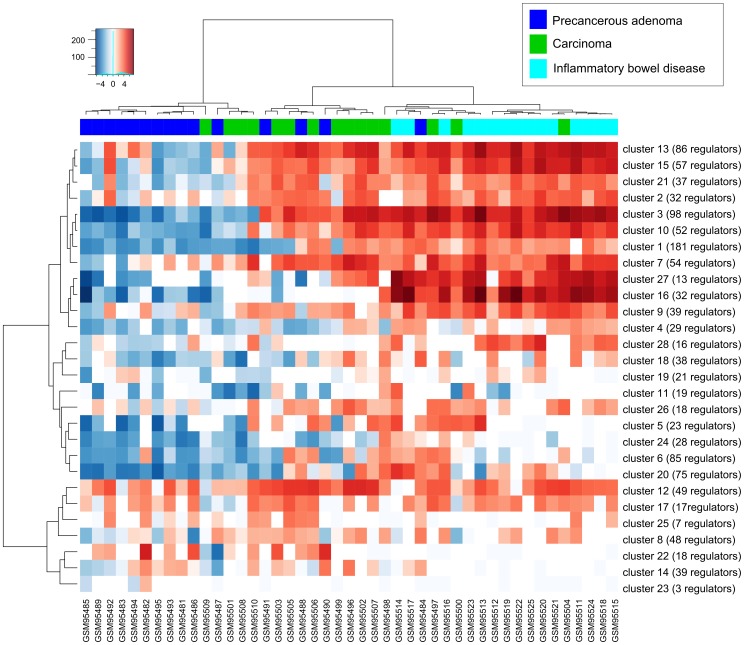
One of the main challenges in modern medicine is to stratify different patient groups in terms of underlying disease molecular mechanisms as to develop more personalized approach to therapy. Here we propose novel method for disease subtyping based on analysis of activated expression regulators on a sample-by-sample basis. Our approach relies on Sub-Network Enrichment Analysis algorithm (SNEA) which identifies gene subnetworks with significant concordant changes in expression between two conditions. Subnetwork consists of central regulator and downstream genes connected by relations extracted from global literature-extracted regulation database. Regulators found in each patient separately are clustered together and assigned activity scores which are used for final patients grouping. We show that our approach performs well compared to other related methods and at the same time provides researchers with complementary level of understanding of pathway-level biology behind a disease by identification of significant expression regulators. We have observed the reasonable grouping of neuromuscular disorders (triggered by structural damage vs triggered by unknown mechanisms), that was not revealed using standard expression profile clustering. For another experiment we were able to suggest the clusters of regulators, responsible for colorectal carcinoma vs adenoma discrimination and identify frequently genetically changed regulators that could be of specific importance for the individual characteristics of cancer development. Proposed approach can be regarded as biologically meaningful feature selection, reducing tens of thousands of genes down to dozens of clusters of regulators. Obtained clusters of regulators make possible to generate valuable biological hypotheses about molecular mechanisms related to a clinical outcome for individual patient.
在现代医学中,主要挑战之一是根据潜在疾病的分子机制对不同的患者群体进行分层,以便为治疗制定更具个性化的方法。在这里,我们提出了一种基于逐个样本分析激活表达调节剂的疾病亚型新方法。我们的方法依赖于子网络富集分析算法(SNEA),该算法可识别两种条件之间表达变化具有显著一致性的基因子网络。子网由中央调节剂和通过从全球文献提取的调控数据库中提取的关系连接的下游基因组成。在每个患者中分别找到的调节剂被聚类在一起,并分配活性评分,用于最终的患者分组。我们表明,与其他相关方法相比,我们的方法表现良好,同时通过识别重要的表达调节剂,为研究人员提供了对疾病背后的通路水平生物学的补充理解。我们观察到了神经肌肉疾病的合理分组(由结构损伤引发与由未知机制引发),这在使用标准表达谱聚类时并未揭示。对于另一个实验,我们能够提示负责结直肠癌与腺瘤区分的调节剂簇,并确定经常发生遗传改变的调节剂,这些调节剂可能对癌症发展的个体特征具有特殊重要性。所提出的方法可以被视为有生物学意义的特征选择,将数万个基因减少到几十个调节剂簇。获得的调节剂簇使得可以生成与个体患者临床结果相关的分子机制的有价值的生物学假设。