Ferragina Paolo, Giancarlo Raffaele, Greco Valentina, Manzini Giovanni, Valiente Gabriel
Dipartimento di Matematica Applicazioni, Università di Palermo, Italy.
BMC Bioinformatics. 2007 Jul 13;8:252. doi: 10.1186/1471-2105-8-252.
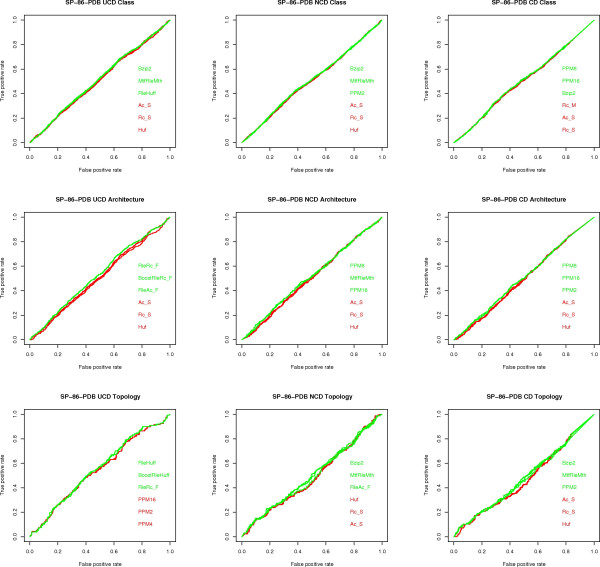
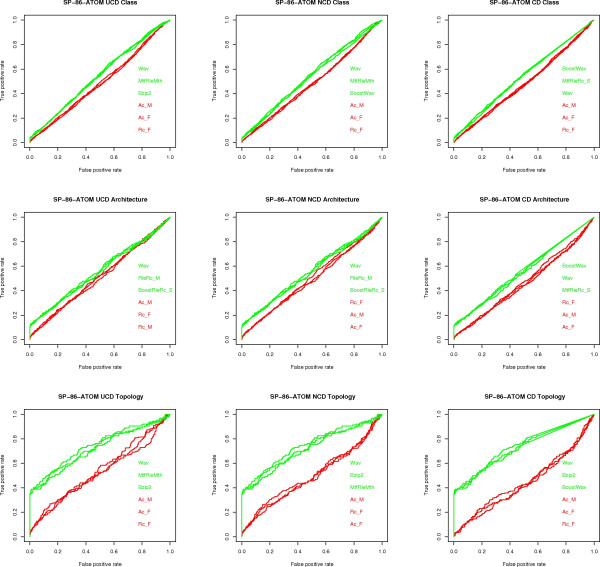
Similarity of sequences is a key mathematical notion for Classification and Phylogenetic studies in Biology. It is currently primarily handled using alignments. However, the alignment methods seem inadequate for post-genomic studies since they do not scale well with data set size and they seem to be confined only to genomic and proteomic sequences. Therefore, alignment-free similarity measures are actively pursued. Among those, USM (Universal Similarity Metric) has gained prominence. It is based on the deep theory of Kolmogorov Complexity and universality is its most novel striking feature. Since it can only be approximated via data compression, USM is a methodology rather than a formula quantifying the similarity of two strings. Three approximations of USM are available, namely UCD (Universal Compression Dissimilarity), NCD (Normalized Compression Dissimilarity) and CD (Compression Dissimilarity). Their applicability and robustness is tested on various data sets yielding a first massive quantitative estimate that the USM methodology and its approximations are of value. Despite the rich theory developed around USM, its experimental assessment has limitations: only a few data compressors have been tested in conjunction with USM and mostly at a qualitative level, no comparison among UCD, NCD and CD is available and no comparison of USM with existing methods, both based on alignments and not, seems to be available.
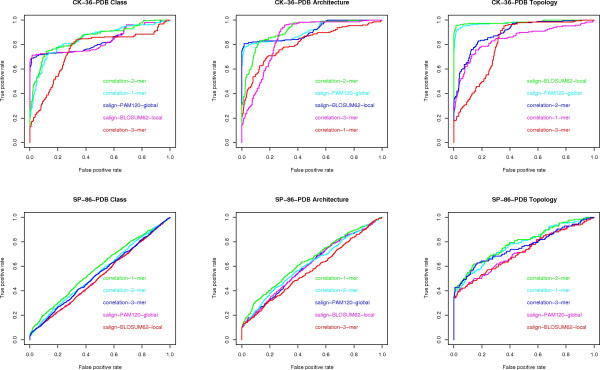
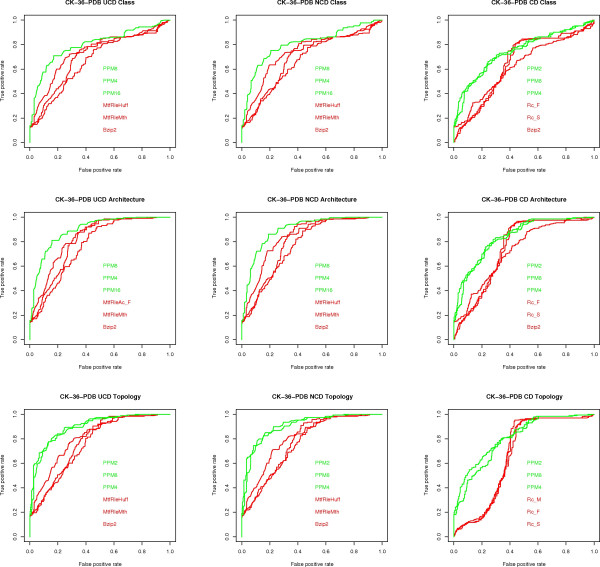
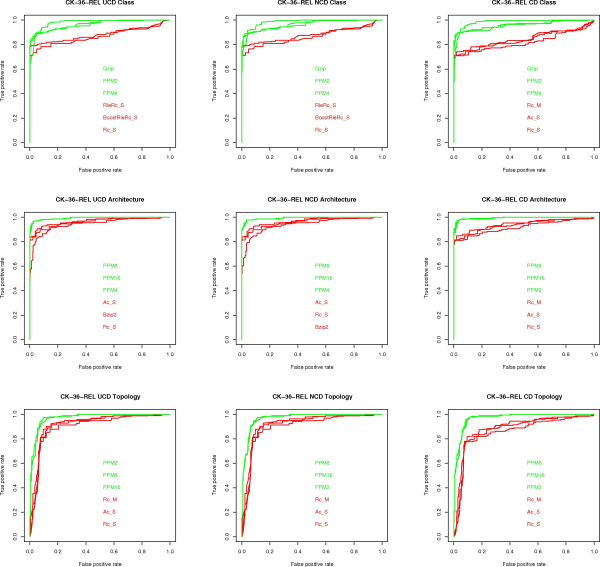
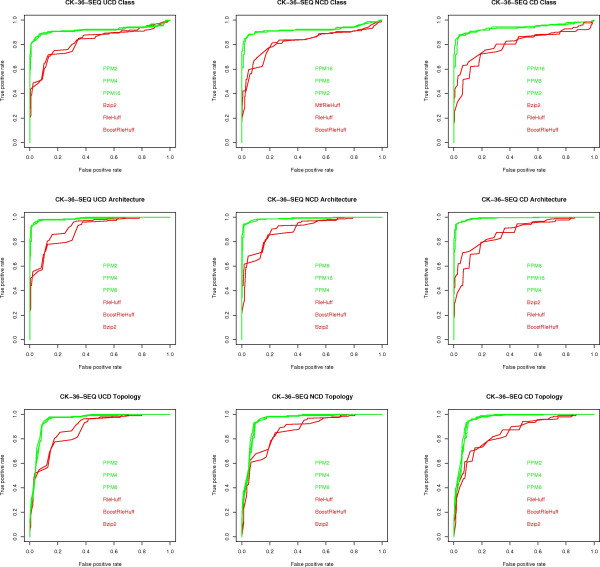
We experimentally test the USM methodology by using 25 compressors, all three of its known approximations and six data sets of relevance to Molecular Biology. This offers the first systematic and quantitative experimental assessment of this methodology, that naturally complements the many theoretical and the preliminary experimental results available. Moreover, we compare the USM methodology both with methods based on alignments and not. We may group our experiments into two sets. The first one, performed via ROC (Receiver Operating Curve) analysis, aims at assessing the intrinsic ability of the methodology to discriminate and classify biological sequences and structures. A second set of experiments aims at assessing how well two commonly available classification algorithms, UPGMA (Unweighted Pair Group Method with Arithmetic Mean) and NJ (Neighbor Joining), can use the methodology to perform their task, their performance being evaluated against gold standards and with the use of well known statistical indexes, i.e., the F-measure and the partition distance. Based on the experiments, several conclusions can be drawn and, from them, novel valuable guidelines for the use of USM on biological data. The main ones are reported next.
UCD and NCD are indistinguishable, i.e., they yield nearly the same values of the statistical indexes we have used, accross experiments and data sets, while CD is almost always worse than both. UPGMA seems to yield better classification results with respect to NJ, i.e., better values of the statistical indexes (10% difference or above), on a substantial fraction of experiments, compressors and USM approximation choices. The compression program PPMd, based on PPM (Prediction by Partial Matching), for generic data and Gencompress for DNA, are the best performers among the compression algorithms we have used, although the difference in performance, as measured by statistical indexes, between them and the other algorithms depends critically on the data set and may not be as large as expected. PPMd used with UCD or NCD and UPGMA, on sequence data is very close, although worse, in performance with the alignment methods (less than 2% difference on the F-measure). Yet, it scales well with data set size and it can work on data other than sequences. In summary, our quantitative analysis naturally complements the rich theory behind USM and supports the conclusion that the methodology is worth using because of its robustness, flexibility, scalability, and competitiveness with existing techniques. In particular, the methodology applies to all biological data in textual format. The software and data sets are available under the GNU GPL at the supplementary material web page.

序列相似性是生物学分类和系统发育研究中的关键数学概念。目前主要通过比对来处理。然而,比对方法对于后基因组研究似乎并不适用,因为它们不能很好地随数据集大小扩展,而且似乎仅局限于基因组和蛋白质组序列。因此,无比对相似性度量方法正被积极探索。其中,通用相似性度量(USM)备受关注。它基于柯尔莫哥洛夫复杂性的深层理论,通用性是其最显著的新颖特征。由于它只能通过数据压缩来近似,USM是一种方法而非量化两个字符串相似性的公式。USM有三种近似方法,即通用压缩差异(UCD)、归一化压缩差异(NCD)和压缩差异(CD)。在各种数据集上对它们的适用性和稳健性进行了测试,得出了首个大规模定量估计,即USM方法及其近似方法具有价值。尽管围绕USM发展了丰富的理论,但其实验评估存在局限性:仅结合少数数据压缩器对USM进行了测试,且大多是定性层面的,没有对UCD、NCD和CD进行比较,也没有将USM与现有方法(包括基于比对和非比对的方法)进行比较。
我们使用25种压缩器、USM的所有三种已知近似方法以及六个与分子生物学相关的数据集,对USM方法进行了实验测试。这为该方法提供了首个系统的定量实验评估,自然地补充了现有的许多理论和初步实验结果。此外,我们将USM方法与基于比对和非比对的方法进行了比较。我们可以将实验分为两组。第一组通过ROC(接收者操作特征曲线)分析进行,旨在评估该方法区分和分类生物序列及结构的内在能力。第二组实验旨在评估两种常用分类算法,即算术平均非加权配对组方法(UPGMA)和邻接法(NJ),如何利用该方法执行其任务,通过与黄金标准对比并使用知名统计指标(即F值和划分距离)来评估它们的性能。基于这些实验,可以得出一些结论,并从中得出关于在生物数据上使用USM的新颖且有价值的指导方针。主要结论如下所述。
UCD和NCD难以区分,即 across实验和数据集,它们得出的我们所使用的统计指标值几乎相同,而CD几乎总是比两者都差。在相当一部分实验、压缩器和USM近似方法选择中,UPGMA相对于NJ似乎能产生更好的分类结果,即统计指标值更好(相差10%或以上)。基于部分匹配预测(PPM)的通用数据压缩程序PPMd和用于DNA的Gencompress,是我们所使用压缩算法中表现最佳的,尽管通过统计指标衡量它们与其他算法在性能上的差异在很大程度上取决于数据集,可能并不像预期的那么大。PPMd与UCD或NCD以及UPGMA一起用于序列数据时,性能与比对方法非常接近,尽管稍差(F值相差不到2%)。然而,它能很好地随数据集大小扩展,并且可以处理序列以外的数据。总之,我们的定量分析自然地补充了USM背后丰富的理论,并支持这样的结论:该方法因其稳健性、灵活性、可扩展性以及与现有技术的竞争力而值得使用。特别是,该方法适用于所有文本格式的生物数据。软件和数据集可在补充材料网页上根据GNU GPL协议获取。