Cohen Philip R
University of Houston Health Center, Houston, Texas, USA.
Orphanet J Rare Dis. 2007 Jul 26;2:34. doi: 10.1186/1750-1172-2-34.
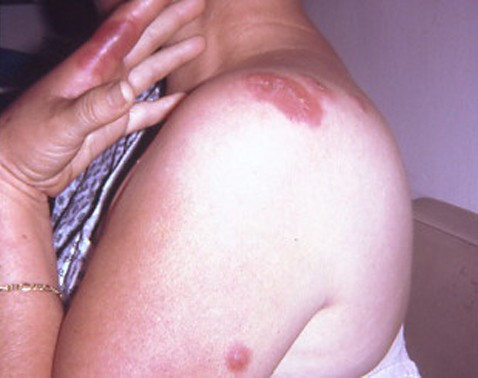
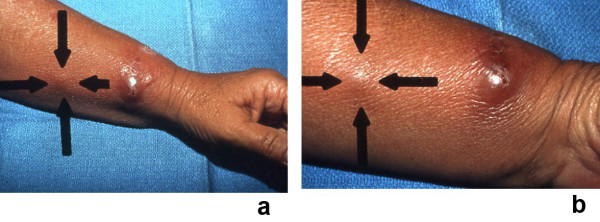
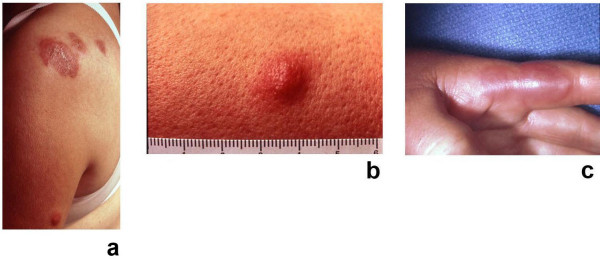
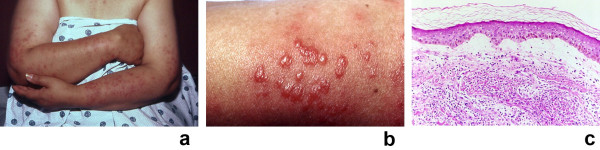
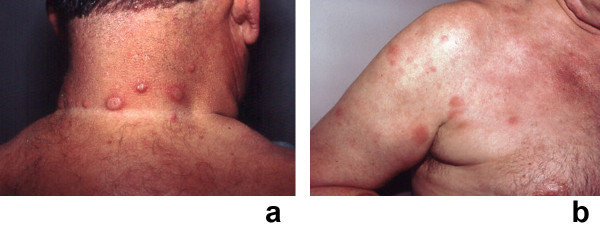
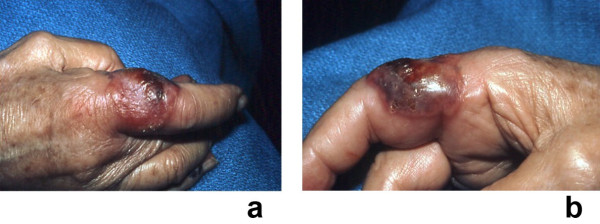
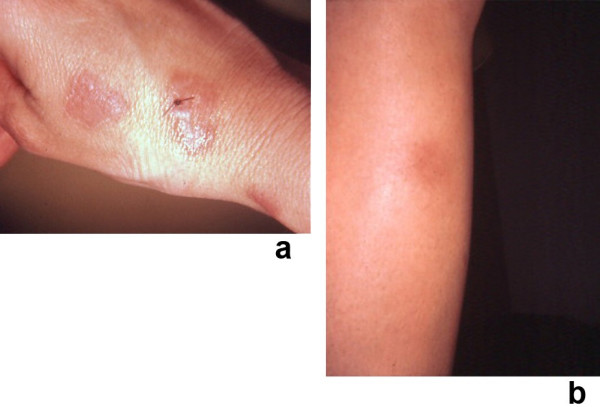
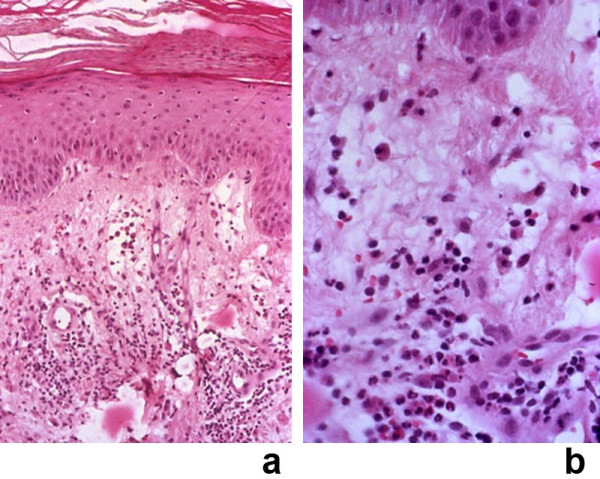
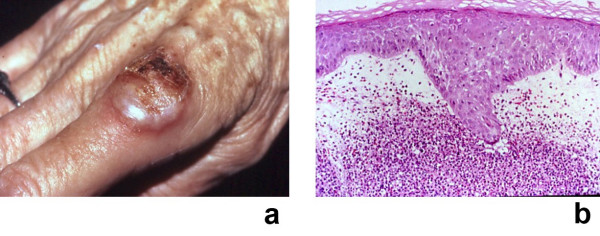
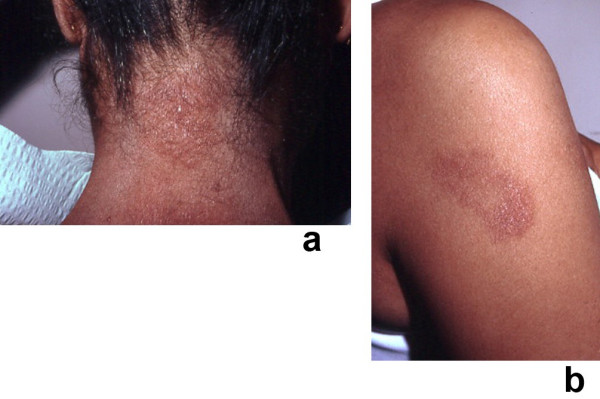
Sweet's syndrome (the eponym for acute febrile neutrophilic dermatosis) is characterized by a constellation of clinical symptoms, physical features, and pathologic findings which include fever, neutrophilia, tender erythematous skin lesions (papules, nodules, and plaques), and a diffuse infiltrate consisting predominantly of mature neutrophils that are typically located in the upper dermis. Several hundreds cases of Sweet's syndrome have been published. Sweet's syndrome presents in three clinical settings: classical (or idiopathic), malignancy-associated, and drug-induced. Classical Sweet's syndrome (CSS) usually presents in women between the age of 30 to 50 years, it is often preceded by an upper respiratory tract infection and may be associated with inflammatory bowel disease and pregnancy. Approximately one-third of patients with CSS experience recurrence of the dermatosis. The malignancy-associated Sweet's syndrome (MASS) can occur as a paraneoplastic syndrome in patients with an established cancer or individuals whose Sweet's syndrome-related hematologic dyscrasia or solid tumor was previously undiscovered; MASS is most commonly related to acute myelogenous leukemia. The dermatosis can precede, follow, or appear concurrent with the diagnosis of the patient's cancer. Hence, MASS can be the cutaneous harbinger of either an undiagnosed visceral malignancy in a previously cancer-free individual or an unsuspected cancer recurrence in an oncology patient. Drug-induced Sweet's syndrome (DISS) most commonly occurs in patients who have been treated with granulocyte-colony stimulating factor, however, other medications may also be associated with DISS. The pathogenesis of Sweet's syndrome may be multifactorial and still remains to be definitively established. Clinical and laboratory evidence suggests that cytokines have an etiologic role. Systemic corticosteroids are the therapeutic gold standard for Sweet's syndrome. After initiation of treatment with systemic corticosteroids, there is a prompt response consisting of dramatic improvement of both the dermatosis-related symptoms and skin lesions. Topical application of high potency corticosteroids or intralesional corticosteroids may be efficacious for treating localized lesions. Other first-line oral systemic agents are potassium iodide and colchicine. Second-line oral systemic agents include indomethacin, clofazimine, cyclosporine, and dapsone. The symptoms and lesions of Sweet's syndrome may resolved spontaneously, without any therapeutic intervention; however, recurrence may follow either spontaneous remission or therapy-induced clinical resolution.
斯威特综合征(急性发热性嗜中性皮病的同义词)的特征是一系列临床症状、体征和病理表现,包括发热、嗜中性粒细胞增多、压痛性红斑皮肤病变(丘疹、结节和斑块),以及主要由成熟嗜中性粒细胞组成的弥漫性浸润,这些细胞通常位于真皮上层。已经发表了数百例斯威特综合征的病例。斯威特综合征有三种临床类型:经典型(或特发性)、恶性肿瘤相关型和药物诱导型。经典斯威特综合征(CSS)通常发生在30至50岁的女性中,常先有上呼吸道感染,可能与炎症性肠病和妊娠有关。约三分之一的CSS患者会出现皮肤病复发。恶性肿瘤相关的斯威特综合征(MASS)可作为副肿瘤综合征出现在已确诊癌症的患者或之前未发现与斯威特综合征相关的血液系统异常或实体瘤的个体中;MASS最常与急性髓系白血病有关。皮肤病可先于、后于或与患者癌症的诊断同时出现。因此,MASS可以是先前无癌症个体中未诊断出的内脏恶性肿瘤的皮肤先兆,也可以是肿瘤患者中未被怀疑的癌症复发的先兆。药物诱导的斯威特综合征(DISS)最常发生在接受粒细胞集落刺激因子治疗的患者中,然而,其他药物也可能与DISS有关。斯威特综合征的发病机制可能是多因素的,仍有待明确确定。临床和实验室证据表明细胞因子起病因作用。全身用皮质类固醇是斯威特综合征的治疗金标准。开始用全身用皮质类固醇治疗后,会迅速出现反应,皮肤病相关症状和皮肤病变都有显著改善。局部应用高效皮质类固醇或皮损内注射皮质类固醇可能对治疗局限性病变有效。其他一线口服全身性药物是碘化钾和秋水仙碱。二线口服全身性药物包括吲哚美辛、氯法齐明、环孢素和氨苯砜。斯威特综合征的症状和病变可能会自发缓解,无需任何治疗干预;然而,复发可能发生在自发缓解或治疗诱导的临床缓解之后。