Chawade Aakash, Bräutigam Marcus, Lindlöf Angelica, Olsson Olof, Olsson Björn
Department of Cell and Molecular Biology, Göteborg University, Box 462, 403 20 Göteborg, Sweden.
BMC Genomics. 2007 Sep 2;8:304. doi: 10.1186/1471-2164-8-304.
With the advent of microarray technology, it has become feasible to identify virtually all genes in an organism that are induced by developmental or environmental changes. However, relying solely on gene expression data may be of limited value if the aim is to infer the underlying genetic networks. Development of computational methods to combine microarray data with other information sources is therefore necessary. Here we describe one such method.
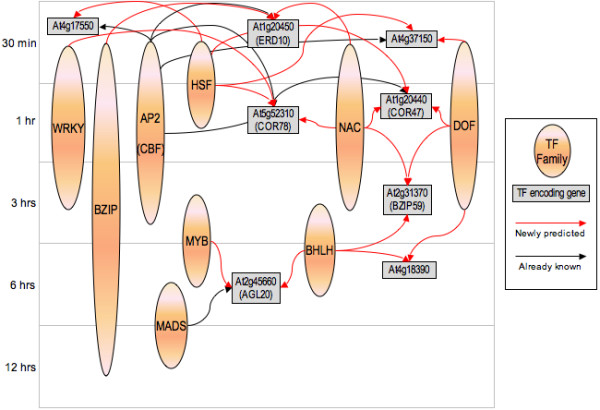
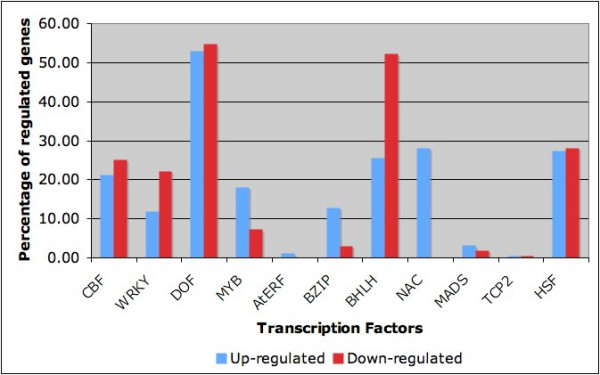
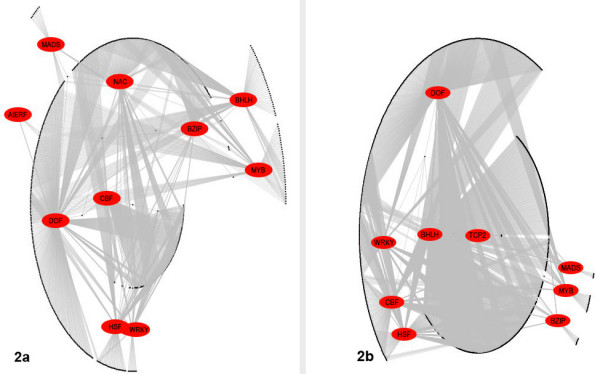
By means of our method, previously published Arabidopsis microarray data from cold acclimated plants at six different time points, promoter motif sequence data extracted from approximately 24,000 Arabidopsis promoters and known transcription factor binding sites were combined to construct a putative genetic regulatory interaction network. The inferred network includes both previously characterised and hitherto un-described regulatory interactions between transcription factor (TF) genes and genes that encode other TFs or other proteins. Part of the obtained transcription factor regulatory network is presented here. More detailed information is available in the additional files.
The rule-based method described here can be used to infer genetic networks by combining data from microarrays, promoter sequences and known promoter binding sites. This method should in principle be applicable to any biological system. We tested the method on the cold acclimation process in Arabidopsis and could identify a more complex putative genetic regulatory network than previously described. However, it should be noted that information on specific binding sites for individual TFs were in most cases not available. Thus, gene targets for the entire TF gene families were predicted. In addition, the networks were built solely by a bioinformatics approach and experimental verifications will be necessary for their final validation. On the other hand, since our method highlights putative novel interactions, more directed experiments could now be performed.
随着微阵列技术的出现,识别生物体中几乎所有由发育或环境变化诱导的基因已变得可行。然而,如果目的是推断潜在的遗传网络,仅依靠基因表达数据可能价值有限。因此,有必要开发将微阵列数据与其他信息源相结合的计算方法。在此,我们描述一种这样的方法。
通过我们的方法,将先前发表的来自六个不同时间点的冷驯化拟南芥微阵列数据、从约24000个拟南芥启动子中提取的启动子基序序列数据以及已知的转录因子结合位点相结合,构建了一个假定的遗传调控相互作用网络。推断出的网络包括转录因子(TF)基因与编码其他TF或其他蛋白质的基因之间先前已表征和迄今未描述的调控相互作用。此处展示了所获得的转录因子调控网络的一部分。更多详细信息见附加文件。
这里描述的基于规则的方法可用于通过结合微阵列数据、启动子序列和已知启动子结合位点来推断遗传网络。该方法原则上应适用于任何生物系统。我们在拟南芥的冷驯化过程中测试了该方法,能够识别出比先前描述的更复杂的假定遗传调控网络。然而,应当指出的是,在大多数情况下,单个TF的特定结合位点信息不可用。因此,预测了整个TF基因家族的基因靶点。此外,这些网络完全通过生物信息学方法构建,其最终验证需要进行实验验证。另一方面,由于我们的方法突出了假定的新相互作用,现在可以进行更具针对性的实验。