Sheng Ying, Engström Pär G, Lenhard Boris
Computational Biology Unit, Bergen Center for Computational Science, University of Bergen, Bergen, Norway.
PLoS One. 2007 Sep 26;2(9):e946. doi: 10.1371/journal.pone.0000946.
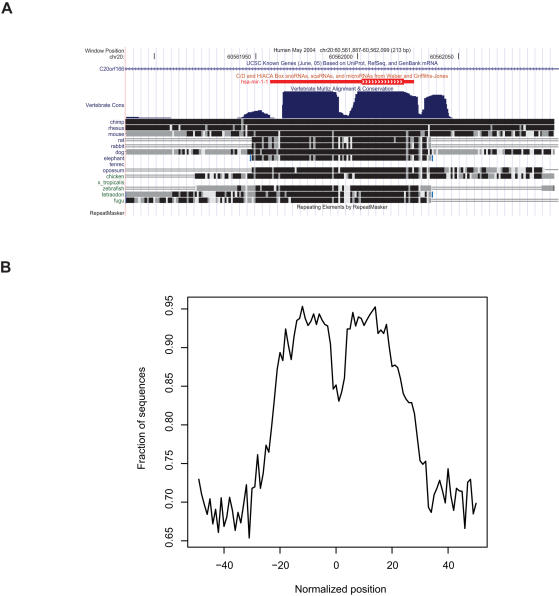
MicroRNAs (miRNAs) are endogenous small noncoding RNA gene products, on average 22 nt long, found in a wide variety of organisms. They play important regulatory roles by targeting mRNAs for degradation or translational repression. There are 377 known mouse miRNAs and 475 known human miRNAs in the May 2007 release of the miRBase database, the majority of which are conserved between the two species. A number of recent reports imply that it is likely that many mammalian miRNAs remain to be discovered. The possibility that there are more of them expressed at lower levels or in more specialized expression contexts calls for the exploitation of genome sequence information to accelerate their discovery.
METHODOLOGY/PRINCIPAL FINDINGS: In this article, we describe a computational method-mirCoS-that uses three support vector machine models sequentially to discover new miRNA candidates in mammalian genomes based on sequence, secondary structure, and conservation. mirCoS can efficiently detect the majority of known miRNAs and predicts an extensive set of hairpin structures based on human-mouse comparisons. In total, 3476 mouse candidates and 3441 human candidates were found. These hairpins are more similar to known miRNAs than to negative controls in several aspects not considered by the prediction algorithm. A significant fraction of predictions is supported by existing expression evidence.
CONCLUSIONS/SIGNIFICANCE: Using a novel approach, mirCoS performs comparably to or better than existing miRNA prediction methods, and contributes a significant number of new candidate miRNAs for experimental verification.
微小RNA(miRNA)是内源性小非编码RNA基因产物,平均长度为22个核苷酸,存在于多种生物中。它们通过靶向mRNA进行降解或翻译抑制发挥重要的调节作用。在2007年5月发布的miRBase数据库中,已知有377种小鼠miRNA和475种人类miRNA,其中大多数在这两个物种之间是保守的。最近的一些报告表明,可能仍有许多哺乳动物miRNA有待发现。存在更多在较低水平或更特殊表达背景下表达的miRNA的可能性,要求利用基因组序列信息来加速它们的发现。
方法/主要发现:在本文中,我们描述了一种计算方法——mirCoS,它依次使用三个支持向量机模型,基于序列、二级结构和保守性在哺乳动物基因组中发现新的miRNA候选物。mirCoS可以有效地检测大多数已知的miRNA,并基于人类-小鼠比较预测大量的发夹结构。总共发现了3476个小鼠候选物和3441个人类候选物。在预测算法未考虑的几个方面,这些发夹与已知miRNA的相似性高于与阴性对照的相似性。相当一部分预测得到了现有表达证据的支持。
结论/意义:使用一种新方法,mirCoS的表现与现有miRNA预测方法相当或更好,并为实验验证贡献了大量新的候选miRNA。