Science for Life Laboratory, Department of Molecular Biosciences, The Wenner-Gren Institute, Stockholm University , Stockholm , Sweden.
Front Bioeng Biotechnol. 2015 Jan 26;3:7. doi: 10.3389/fbioe.2015.00007. eCollection 2015.
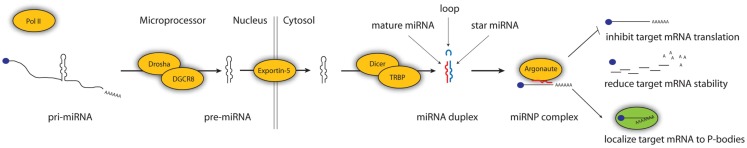
Next-generation sequencing now for the first time allows researchers to gage the depth and variation of entire transcriptomes. However, now as rare transcripts can be detected that are present in cells at single copies, more advanced computational tools are needed to accurately annotate and profile them. microRNAs (miRNAs) are 22 nucleotide small RNAs (sRNAs) that post-transcriptionally reduce the output of protein coding genes. They have established roles in numerous biological processes, including cancers and other diseases. During miRNA biogenesis, the sRNAs are sequentially cleaved from precursor molecules that have a characteristic hairpin RNA structure. The vast majority of new miRNA genes that are discovered are mined from small RNA sequencing (sRNA-seq), which can detect more than a billion RNAs in a single run. However, given that many of the detected RNAs are degradation products from all types of transcripts, the accurate identification of miRNAs remain a non-trivial computational problem. Here, we review the tools available to predict animal miRNAs from sRNA sequencing data. We present tools for generalist and specialist use cases, including prediction from massively pooled data or in species without reference genome. We also present wet-lab methods used to validate predicted miRNAs, and approaches to computationally benchmark prediction accuracy. For each tool, we reference validation experiments and benchmarking efforts. Last, we discuss the future of the field.
下一代测序技术现在首次允许研究人员衡量整个转录组的深度和变化。然而,现在由于可以检测到在单个细胞中存在的稀有转录本,因此需要更先进的计算工具来准确注释和分析它们。 microRNAs (miRNAs) 是 22 个核苷酸的小 RNA (sRNA),可在后转录水平降低蛋白质编码基因的产物。它们在许多生物学过程中发挥作用,包括癌症和其他疾病。在 miRNA 生物发生过程中,sRNA 从前体分子中顺序切割,前体分子具有特征性发夹 RNA 结构。从小 RNA 测序 (sRNA-seq) 中挖掘出的大多数新的 miRNA 基因,单次运行就可以检测超过十亿个 RNA。然而,鉴于许多检测到的 RNA 是来自各种转录本的降解产物,miRNA 的准确识别仍然是一个非平凡的计算问题。在这里,我们回顾了可用于从 sRNA 测序数据中预测动物 miRNA 的工具。我们介绍了通用和专业用例的工具,包括从大量混合数据或没有参考基因组的物种中进行预测。我们还介绍了用于验证预测 miRNA 的湿实验室方法,以及计算预测准确性的方法。对于每个工具,我们都参考了验证实验和基准测试工作。最后,我们讨论了该领域的未来。