Opavsky Rene, Wang Shu-Huei, Trikha Prashant, Raval Aparna, Huang Yuan, Wu Yue-Zhong, Rodriguez Benjamin, Keller Benjamin, Liyanarachchi Sandya, Wei Guo, Davuluri Ramana V, Weinstein Michael, Felsher Dean, Ostrowski Michael, Leone Gustavo, Plass Christoph
Human Cancer Genetics Program, Department of Molecular Virology, Immunology and Medical Genetics, The Ohio State University, Columbus, Ohio, USA.
PLoS Genet. 2007 Sep;3(9):1757-69. doi: 10.1371/journal.pgen.0030167. Epub 2007 Aug 16.
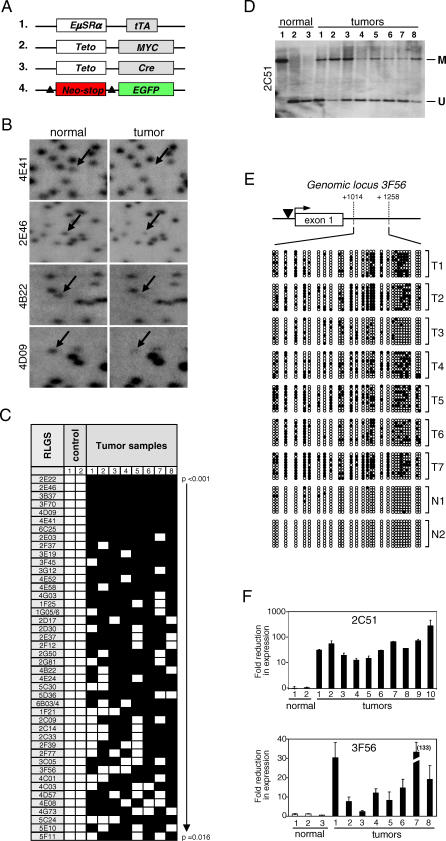
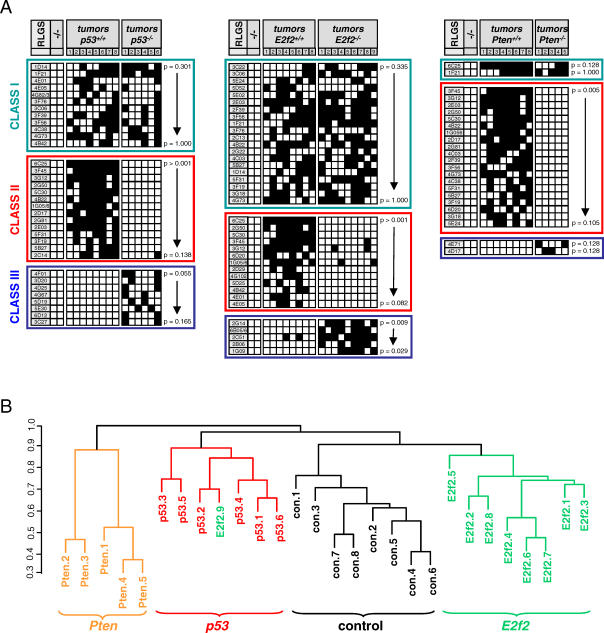
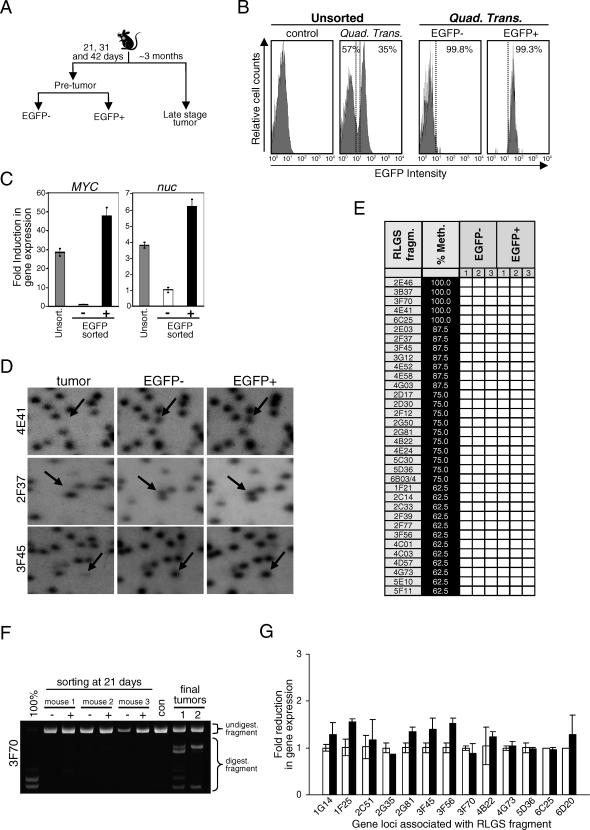
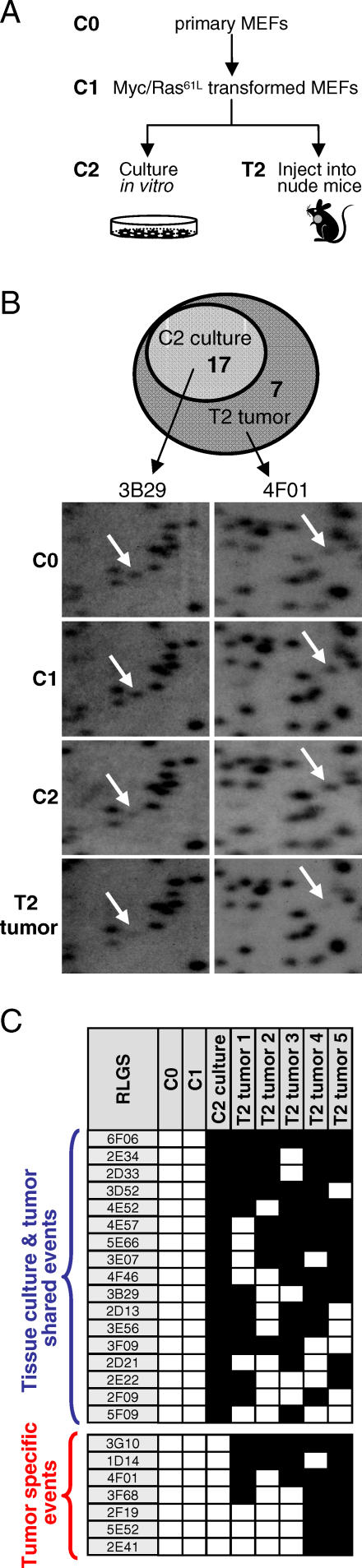
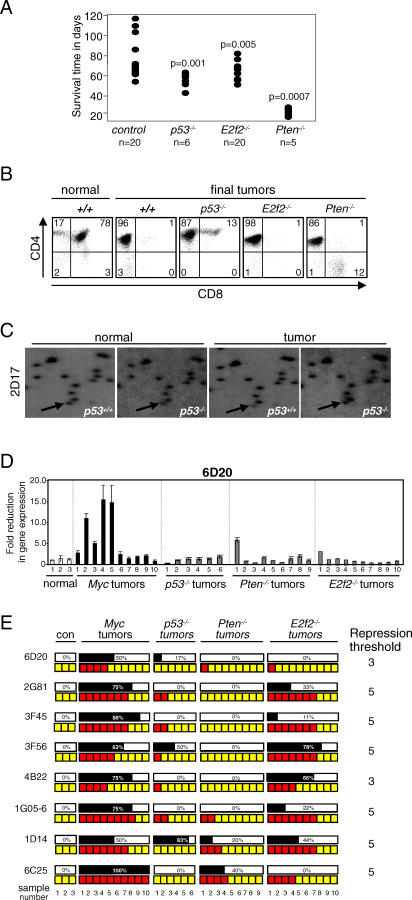
Hypermethylation of CpG islands is a common epigenetic alteration associated with cancer. Global patterns of hypermethylation are tumor-type specific and nonrandom. The biological significance and the underlying mechanisms of tumor-specific aberrant promoter methylation remain unclear, but some evidence suggests that this specificity involves differential sequence susceptibilities, the targeting of DNA methylation activity to specific promoter sequences, or the selection of rare DNA methylation events during disease progression. Using restriction landmark genomic scanning on samples derived from tissue culture and in vivo models of T cell lymphomas, we found that MYC overexpression gave rise to a specific signature of CpG island hypermethylation. This signature reflected gene transcription profiles and was detected only in advanced stages of disease. The further inactivation of the Pten, p53, and E2f2 tumor suppressors in MYC-induced lymphomas resulted in distinct and diagnostic CpG island methylation signatures. Our data suggest that tumor-specific DNA methylation in lymphomas arises as a result of the selection of rare DNA methylation events during the course of tumor development. This selection appears to be driven by the genetic configuration of tumor cells, providing experimental evidence for a causal role of DNA hypermethylation in tumor progression and an explanation for the tremendous epigenetic heterogeneity observed in the evolution of human cancers. The ability to predict genome-wide epigenetic silencing based on relatively few genetic alterations will allow for a more complete classification of tumors and understanding of tumor cell biology.
CpG岛的高甲基化是一种与癌症相关的常见表观遗传改变。高甲基化的整体模式具有肿瘤类型特异性且是非随机的。肿瘤特异性异常启动子甲基化的生物学意义和潜在机制仍不清楚,但一些证据表明,这种特异性涉及不同的序列敏感性、DNA甲基化活性对特定启动子序列的靶向作用,或疾病进展过程中罕见DNA甲基化事件的选择。通过对来自组织培养和T细胞淋巴瘤体内模型的样本进行限制性内切酶基因组扫描,我们发现MYC过表达导致了CpG岛高甲基化的特定特征。这种特征反映了基因转录谱,并且仅在疾病晚期才被检测到。在MYC诱导的淋巴瘤中,Pten、p53和E2f2肿瘤抑制因子的进一步失活导致了独特的、可诊断的CpG岛甲基化特征。我们的数据表明,淋巴瘤中肿瘤特异性DNA甲基化是肿瘤发展过程中罕见DNA甲基化事件选择的结果。这种选择似乎是由肿瘤细胞的基因构型驱动的,为DNA高甲基化在肿瘤进展中的因果作用提供了实验证据,并解释了在人类癌症演变过程中观察到的巨大表观遗传异质性。基于相对较少的基因改变来预测全基因组表观遗传沉默的能力,将有助于对肿瘤进行更完整的分类,并理解肿瘤细胞生物学。