Wu Randy Z, Chaivorapol Christina, Zheng Jiashun, Li Hao, Liang Shoudan
Department of Biochemistry and Biophysics, UCSF, 1700 4th Street, San Francisco, CA 94143-2542, USA.
BMC Bioinformatics. 2007 Oct 17;8:399. doi: 10.1186/1471-2105-8-399.
The precision of transcriptional regulation is made possible by the specificity of physical interactions between transcription factors and their cognate binding sites on DNA. A major challenge is to decipher transcription factor binding sites from sequence and functional genomic data using computational means. While current methods can detect strong binding sites, they are less sensitive to degenerate motifs.
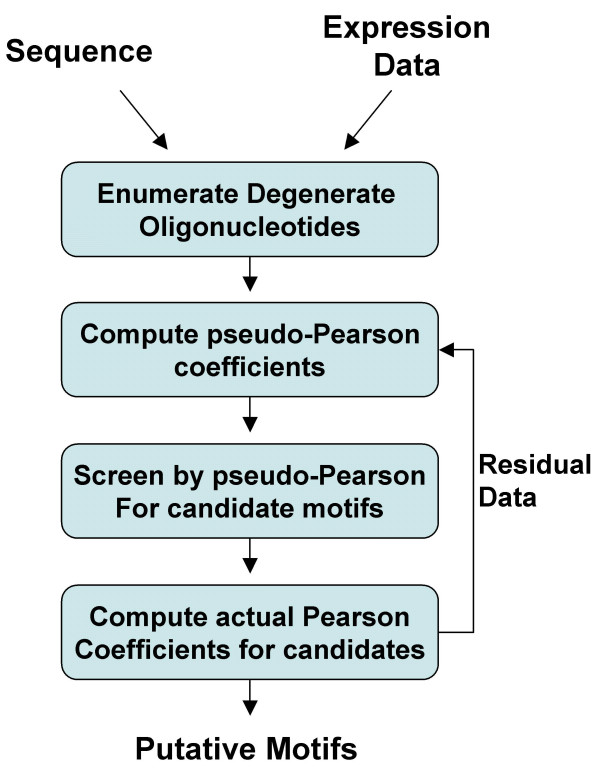
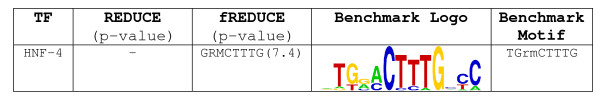
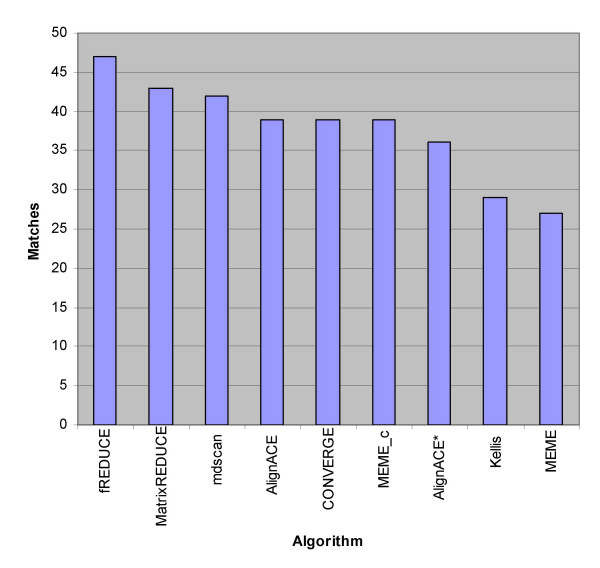
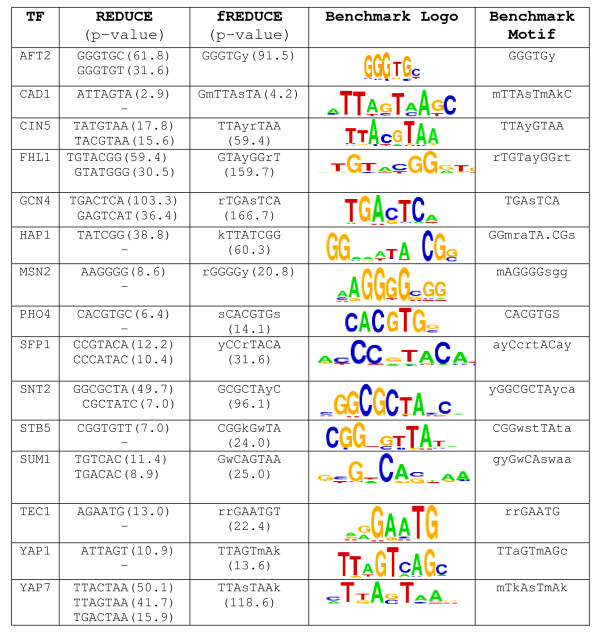
We present fREDUCE, a computational method specialized for the detection of weak or degenerate binding motifs from gene expression or ChIP-chip data. fREDUCE is built upon the widely applied program REDUCE, which elicits motifs by global statistical correlation of motif counts with expression data. fREDUCE introduces several algorithmic refinements that allow efficient exhaustive searches of oligonucleotides with a specified number of degenerate IUPAC symbols. On yeast ChIP-chip benchmarks, fREDUCE correctly identified motifs and their degeneracies with accuracies greater than its predecessor REDUCE as well as other known motif-finding programs. We have also used fREDUCE to make novel motif predictions for transcription factors with poorly characterized binding sites.
We demonstrate that fREDUCE is a valuable tool for the prediction of degenerate transcription factor binding sites, especially from array datasets with weak signals that may elude other motif detection methods.
转录调节的精确性是由转录因子与其在DNA上的同源结合位点之间物理相互作用的特异性实现的。一个主要挑战是使用计算方法从序列和功能基因组数据中破译转录因子结合位点。虽然目前的方法能够检测到强结合位点,但它们对简并基序的敏感性较低。
我们提出了fREDUCE,一种专门用于从基因表达或芯片数据中检测弱或简并结合基序的计算方法。fREDUCE基于广泛应用的程序REDUCE构建,REDUCE通过基序计数与表达数据的全局统计相关性来引出基序。fREDUCE引入了几种算法改进,允许对具有指定数量简并IUPAC符号的寡核苷酸进行高效的穷举搜索。在酵母芯片基准测试中,fREDUCE正确识别了基序及其简并性,其准确率高于其前身REDUCE以及其他已知的基序查找程序。我们还使用fREDUCE对结合位点特征不明确的转录因子进行了新的基序预测。
我们证明fREDUCE是预测简并转录因子结合位点的一个有价值的工具,特别是从可能避开其他基序检测方法的弱信号阵列数据集中。