Gu Jin, Fu Hu, Zhang Xuegong, Li Yanda
Bioinformatics Division, TNLIST and Department of Automation, Tsinghua University, Beijing 100084, China.
BMC Bioinformatics. 2007 Nov 8;8:432. doi: 10.1186/1471-2105-8-432.
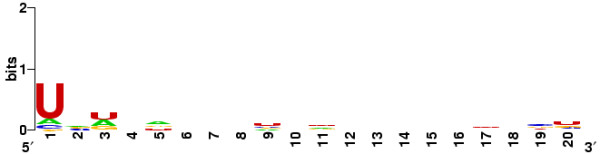
MicroRNAs (miRNAs) are a class of endogenous regulatory small RNAs which play an important role in posttranscriptional regulations by targeting mRNAs for cleavage or translational repression. The base-pairing between the 5'-end of miRNA and the target mRNA 3'-UTRs is essential for the miRNA:mRNA recognition. Recent studies show that many seed matches in 3'-UTRs, which are fully complementary to miRNA 5'-ends, are highly conserved. Based on these features, a two-stage strategy can be implemented to achieve the de novo identification of miRNAs by requiring the complete base-pairing between the 5'-end of miRNA candidates and the potential seed matches in 3'-UTRs.
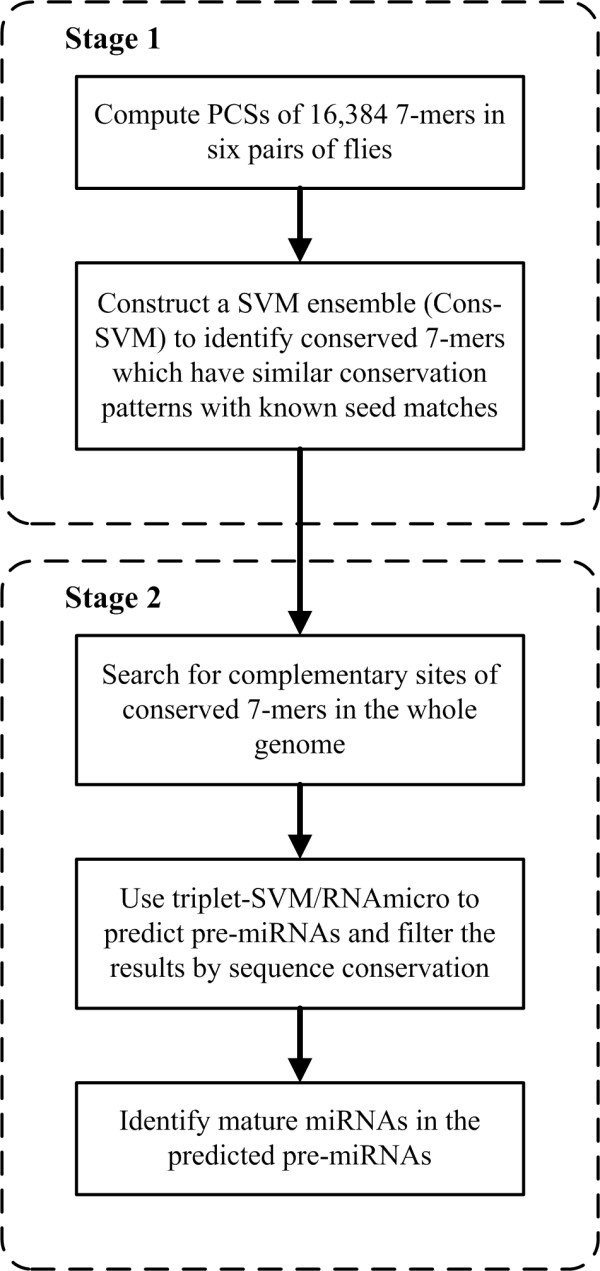
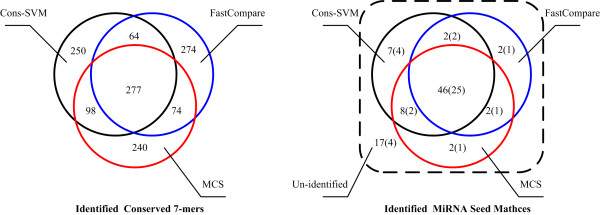
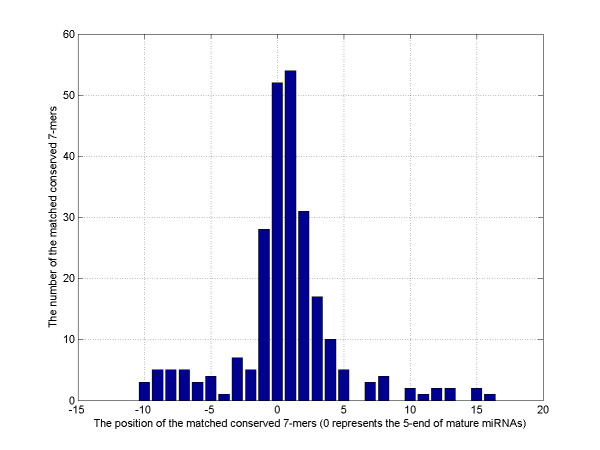
We presented a new method, which combined multiple pairwise conservation information, to identify the frequently-occurred and conserved 7-mers in 3'-UTRs. A pairwise conservation score (PCS) was introduced to describe the conservation of all 7-mers in 3'-UTRs between any two Drosophila species. Using PCSs computed from 6 pairs of flies, we developed a support vector machine (SVM) classifier ensemble, named Cons-SVM and identified 689 conserved 7-mers including 63 seed matches covering 32 out of 38 known miRNA families in the reference dataset. In the second stage, we searched for 90 nt conserved stem-loop regions containing the complementary sequences to the identified 7-mers and used the previously published miRNA prediction software to analyze these stem-loops. We predicted 47 miRNA candidates in the genome-wide screen.
Cons-SVM takes advantage of the independent evolutionary information from the 6 pairs of flies and shows high sensitivity in identifying seed matches in 3'-UTRs. Combining the multiple pairwise conservation information by the machine learning approach, we finally identified 47 miRNA candidates in D. melanogaster.
微小RNA(miRNA)是一类内源性调控小RNA,通过靶向mRNA进行切割或翻译抑制,在转录后调控中发挥重要作用。miRNA 5'端与靶mRNA 3'非翻译区(UTR)之间的碱基配对对于miRNA与mRNA的识别至关重要。最近的研究表明,3'UTR中许多与miRNA 5'端完全互补的种子匹配序列高度保守。基于这些特征,可以实施两阶段策略,通过要求候选miRNA 5'端与3'UTR中的潜在种子匹配序列完全碱基配对,来实现miRNA的从头鉴定。
我们提出了一种结合多个成对保守信息的新方法,用于鉴定在mRNA 3'UTR中频繁出现且保守的7聚体。引入了成对保守得分(PCS)来描述任意两个果蝇物种之间3'UTR中所有7聚体的保守性。利用从6对果蝇计算得到的PCS值,我们开发了一个支持向量机(SVM)分类器集成,命名为Cons-SVM,并鉴定出689个保守的7聚体,其中包括63个种子匹配序列,涵盖了参考数据集中38个已知miRNA家族中的32个。在第二阶段,我们搜索了包含与鉴定出的7聚体互补序列的90 nt保守茎环区域,并使用先前发表的miRNA预测软件分析这些茎环。我们在全基因组筛选中预测了47个miRNA候选序列。
Cons-SVM利用了6对果蝇的独立进化信息,在鉴定3'UTR中的种子匹配序列方面表现出高灵敏度。通过机器学习方法结合多个成对保守信息,我们最终在黑腹果蝇中鉴定出47个miRNA候选序列。