Bernardes Juliana S, Dávila Alberto M R, Costa Vítor S, Zaverucha Gerson
COPPE, Programa de Engenharia de Sistemas e Computação, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil.
BMC Bioinformatics. 2007 Nov 9;8:435. doi: 10.1186/1471-2105-8-435.
Remote homology detection is a challenging problem in Bioinformatics. Arguably, profile Hidden Markov Models (pHMMs) are one of the most successful approaches in addressing this important problem. pHMM packages present a relatively small computational cost, and perform particularly well at recognizing remote homologies. This raises the question of whether structural alignments could impact the performance of pHMMs trained from proteins in the Twilight Zone, as structural alignments are often more accurate than sequence alignments at identifying motifs and functional residues. Next, we assess the impact of using structural alignments in pHMM performance.
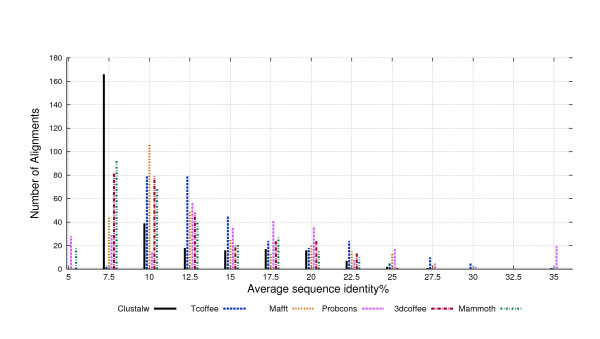
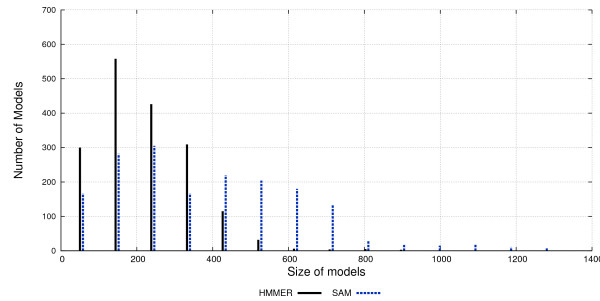
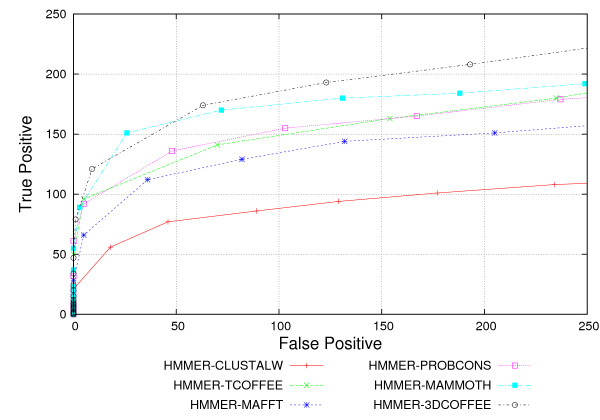
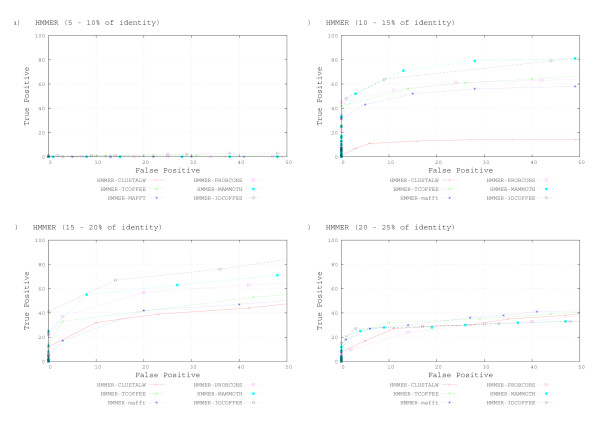
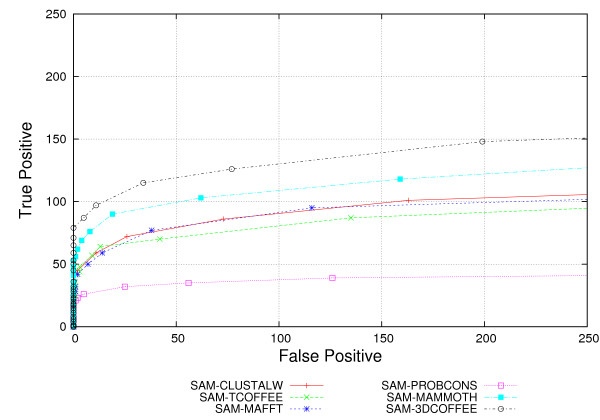
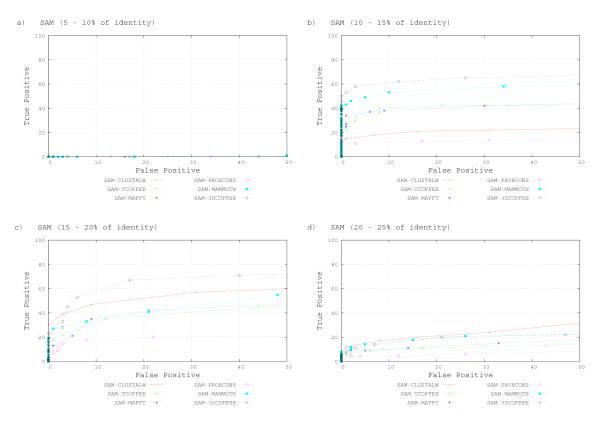
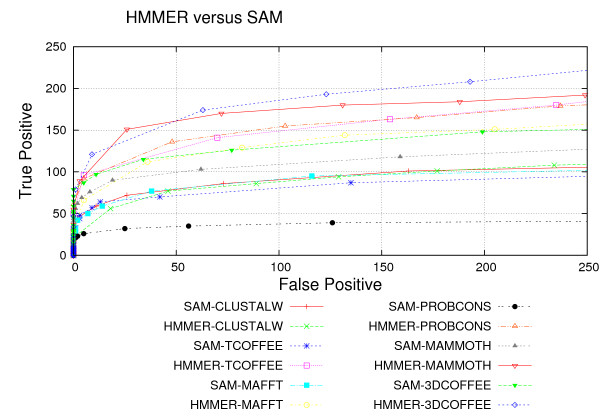
We used the SCOP database to perform our experiments. Structural alignments were obtained using the 3DCOFFEE and MAMMOTH-mult tools; sequence alignments were obtained using CLUSTALW, TCOFFEE, MAFFT and PROBCONS. We performed leave-one-family-out cross-validation over super-families. Performance was evaluated through ROC curves and paired two tailed t-test.
We observed that pHMMs derived from structural alignments performed significantly better than pHMMs derived from sequence alignment in low-identity regions, mainly below 20%. We believe this is because structural alignment tools are better at focusing on the important patterns that are more often conserved through evolution, resulting in higher quality pHMMs. On the other hand, sensitivity of these tools is still quite low for these low-identity regions. Our results suggest a number of possible directions for improvements in this area.
远程同源性检测是生物信息学中的一个具有挑战性的问题。可以说,轮廓隐马尔可夫模型(pHMM)是解决这一重要问题最成功的方法之一。pHMM软件包的计算成本相对较低,在识别远程同源性方面表现尤其出色。这就提出了一个问题,即结构比对是否会影响从处于“黄昏区”的蛋白质训练得到的pHMM的性能,因为在识别基序和功能残基方面,结构比对通常比序列比对更准确。接下来,我们评估在pHMM性能中使用结构比对的影响。
我们使用SCOP数据库进行实验。使用3DCOFFEE和MAMMOTH-mult工具获得结构比对;使用CLUSTALW、TCOFFEE、MAFFT和PROBCONS获得序列比对。我们对超家族进行留一法交叉验证。通过ROC曲线和配对双尾t检验评估性能。
我们观察到,在低同一性区域,即主要低于20%的区域,从结构比对得到的pHMM比从序列比对得到的pHMM表现明显更好。我们认为这是因为结构比对工具更善于关注那些在进化过程中更常保守的重要模式,从而产生更高质量的pHMM。另一方面,这些工具在这些低同一性区域的敏感性仍然相当低。我们的结果为该领域的改进提出了一些可能的方向。