Srivastava Prashant K, Desai Dhwani K, Nandi Soumyadeep, Lynn Andrew M
School of Information Technology, Jawaharlal Nehru University, New Delhi, India.
BMC Bioinformatics. 2007 Mar 27;8:104. doi: 10.1186/1471-2105-8-104.
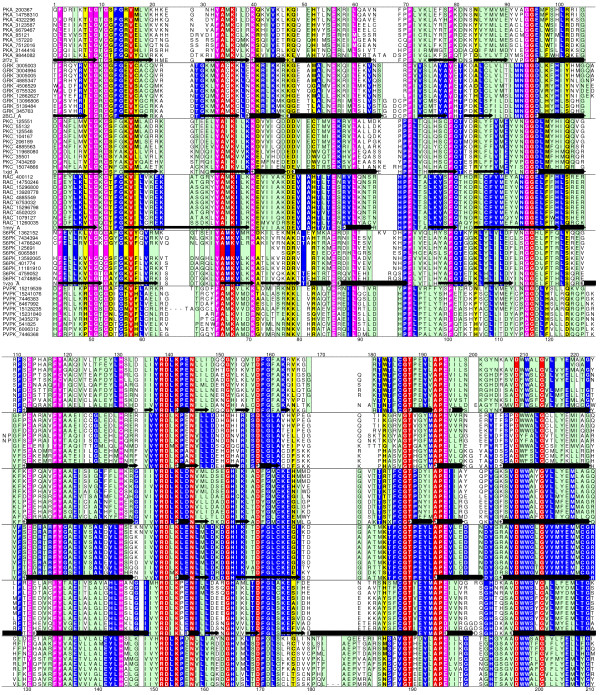
Profile Hidden Markov Models (HMM) are statistical representations of protein families derived from patterns of sequence conservation in multiple alignments and have been used in identifying remote homologues with considerable success. These conservation patterns arise from fold specific signals, shared across multiple families, and function specific signals unique to the families. The availability of sequences pre-classified according to their function permits the use of negative training sequences to improve the specificity of the HMM, both by optimizing the threshold cutoff and by modifying emission probabilities to minimize the influence of fold-specific signals. A protocol to generate family specific HMMs is described that first constructs a profile HMM from an alignment of the family's sequences and then uses this model to identify sequences belonging to other classes that score above the default threshold (false positives). Ten-fold cross validation is used to optimise the discrimination threshold score for the model. The advent of fast multiple alignment methods enables the use of the profile alignments to align the true and false positive sequences, and the resulting alignments are used to modify the emission probabilities in the original model.
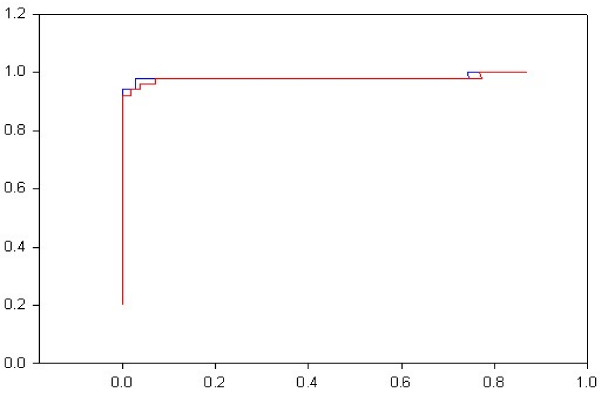
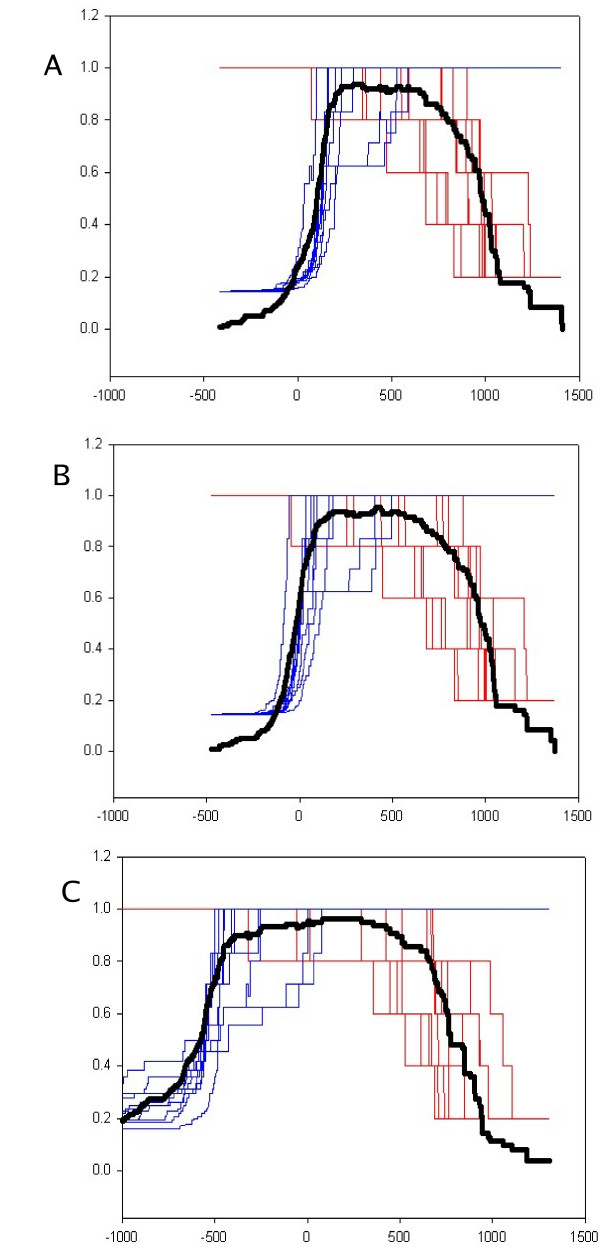
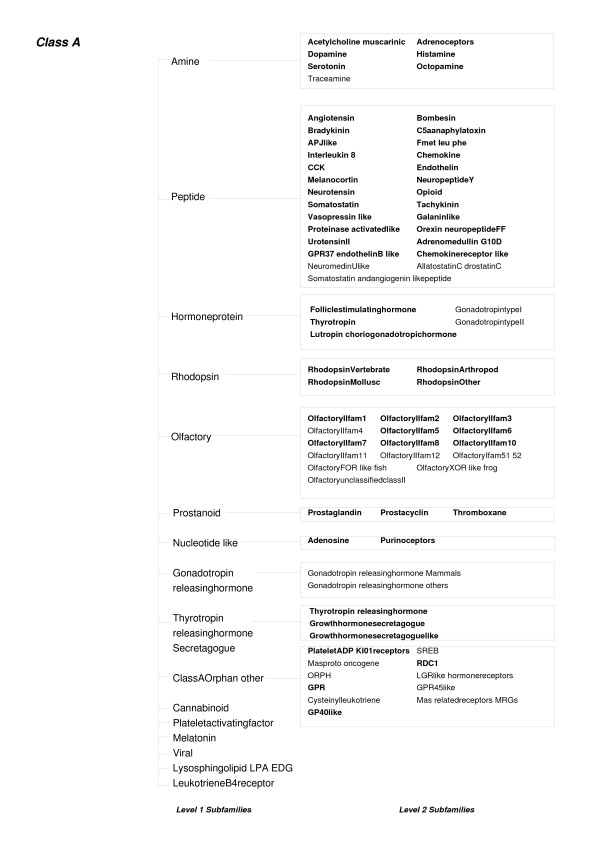
The protocol, called HMM-ModE, was validated on a set of sequences belonging to six sub-families of the AGC family of kinases. These sequences have an average sequence similarity of 63% among the group though each sub-group has a different substrate specificity. The optimisation of discrimination threshold, by using negative sequences scored against the model improves specificity in test cases from an average of 21% to 98%. Further discrimination by the HMM after modifying model probabilities using negative training sequences is provided in a few cases, the average specificity rising to 99%. Similar improvements were obtained with a sample of G-Protein coupled receptors sub-classified with respect to their substrate specificity, though the average sequence identity across the sub-families is just 20.6%. The protocol is applied in a high-throughput classification exercise on protein kinases.
The protocol has the potential to maximise the contributions of discriminating residues to classify proteins based on their molecular function, using pre-classified positive and negative sequence training data. The high specificity of the method, and increasing availability of pre-classified sequence data holds the potential for its application in sequence annotation.
轮廓隐马尔可夫模型(HMM)是基于多序列比对中的序列保守模式得到的蛋白质家族的统计表示,已成功用于识别远源同源物。这些保守模式源于多个家族共有的折叠特异性信号以及各家族特有的功能特异性信号。根据功能预先分类的序列的可用性允许使用负训练序列来提高HMM的特异性,方法是优化阈值截止值以及修改发射概率以最小化折叠特异性信号的影响。本文描述了一种生成家族特异性HMM的方案,该方案首先从家族序列的比对构建一个轮廓HMM,然后使用该模型识别得分高于默认阈值(假阳性)的属于其他类别的序列。采用十折交叉验证来优化模型的判别阈值分数。快速多序列比对方法的出现使得能够使用轮廓比对来比对真阳性和假阳性序列,并且所得比对用于修改原始模型中的发射概率。
该方案称为HMM-ModE,在一组属于AGC激酶家族六个亚家族的序列上进行了验证。这些序列在组内平均序列相似性为63%,尽管每个亚组具有不同的底物特异性。通过使用针对模型评分的负序列来优化判别阈值,可将测试案例中的特异性从平均21%提高到98%。在少数情况下,使用负训练序列修改模型概率后,HMM进一步进行判别,平均特异性提高到99%。对于根据底物特异性进行亚分类的G蛋白偶联受体样本也获得了类似的改进,尽管亚家族之间的平均序列同一性仅为20.6%。该方案应用于蛋白质激酶的高通量分类实验。
该方案有潜力利用预先分类的正序列和负序列训练数据,最大化区分性残基对基于分子功能对蛋白质进行分类的贡献。该方法的高特异性以及预先分类的序列数据可用性的增加,使其有潜力应用于序列注释。