Levitsky Victor G, Ignatieva Elena V, Ananko Elena A, Turnaev Igor I, Merkulova Tatyana I, Kolchanov Nikolay A, Hodgman T C
Institute of Cytology and Genetics SB RAS, Novosibirsk, 630090, Russia.
BMC Bioinformatics. 2007 Dec 19;8:481. doi: 10.1186/1471-2105-8-481.
Reliable transcription factor binding site (TFBS) prediction methods are essential for computer annotation of large amount of genome sequence data. However, current methods to predict TFBSs are hampered by the high false-positive rates that occur when only sequence conservation at the core binding-sites is considered.
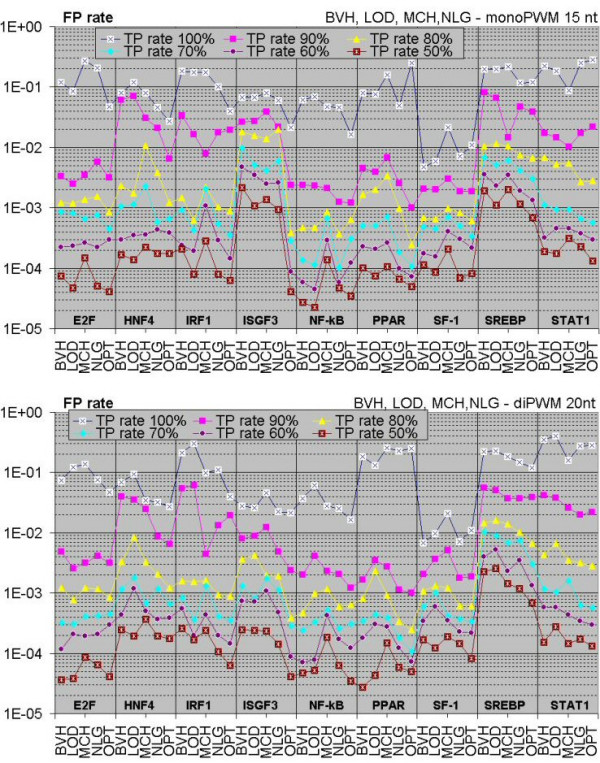
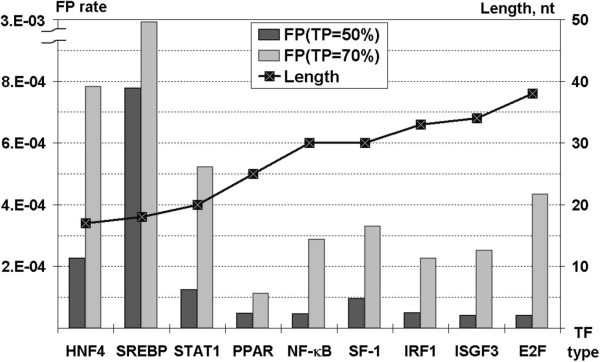
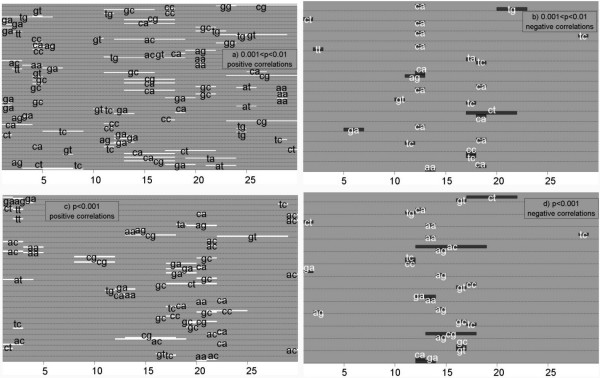
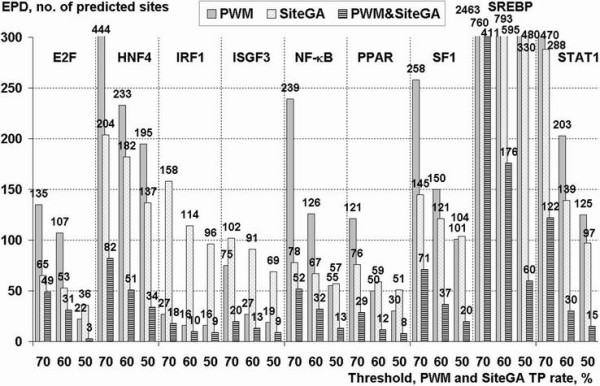
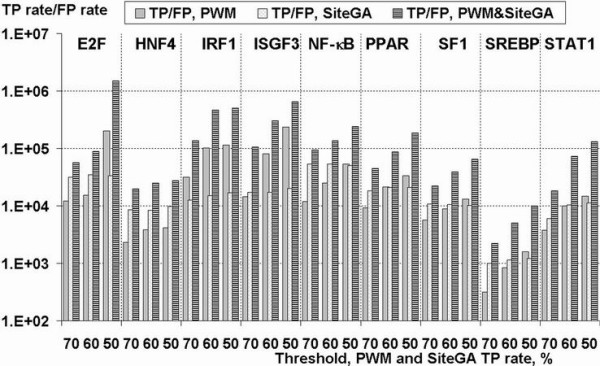
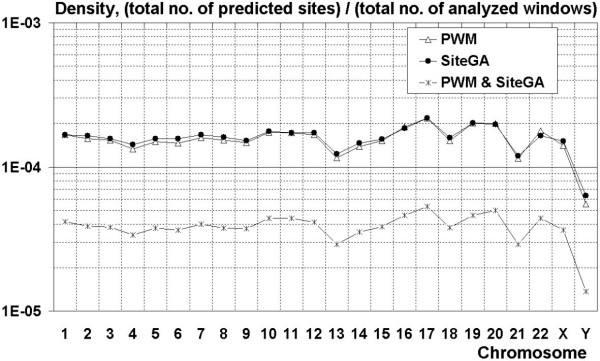
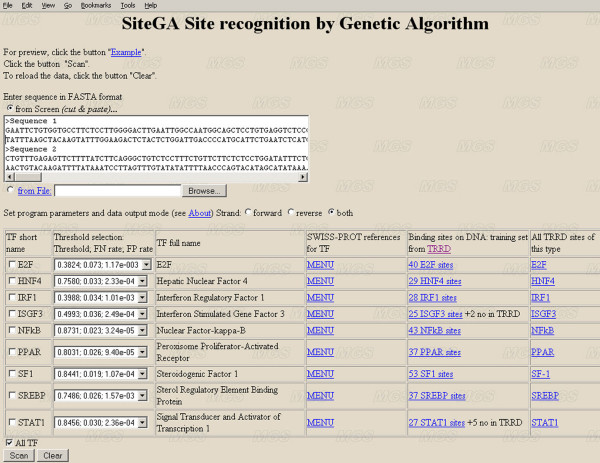
To improve this situation, we have quantified the performance of several Position Weight Matrix (PWM) algorithms, using exhaustive approaches to find their optimal length and position. We applied these approaches to bio-medically important TFBSs involved in the regulation of cell growth and proliferation as well as in inflammatory, immune, and antiviral responses (NF-kappaB, ISGF3, IRF1, STAT1), obesity and lipid metabolism (PPAR, SREBP, HNF4), regulation of the steroidogenic (SF-1) and cell cycle (E2F) genes expression. We have also gained extra specificity using a method, entitled SiteGA, which takes into account structural interactions within TFBS core and flanking regions, using a genetic algorithm (GA) with a discriminant function of locally positioned dinucleotide (LPD) frequencies. To ensure a higher confidence in our approach, we applied resampling-jackknife and bootstrap tests for the comparison, it appears that, optimized PWM and SiteGA have shown similar recognition performances. Then we applied SiteGA and optimized PWMs (both separately and together) to sequences in the Eukaryotic Promoter Database (EPD). The resulting SiteGA recognition models can now be used to search sequences for BSs using the web tool, SiteGA. Analysis of dependencies between close and distant LPDs revealed by SiteGA models has shown that the most significant correlations are between close LPDs, and are generally located in the core (footprint) region. A greater number of less significant correlations are mainly between distant LPDs, which spanned both core and flanking regions. When SiteGA and optimized PWM models were applied together, this substantially reduced false positives at least at higher stringencies.
Based on this analysis, SiteGA adds substantial specificity even to optimized PWMs and may be considered for large-scale genome analysis. It adds to the range of techniques available for TFBS prediction, and EPD analysis has led to a list of genes which appear to be regulated by the above TFs.
可靠的转录因子结合位点(TFBS)预测方法对于大量基因组序列数据的计算机注释至关重要。然而,当前预测TFBS的方法受到仅考虑核心结合位点处的序列保守性时出现的高假阳性率的阻碍。
为改善这种情况,我们使用穷举方法来找到其最佳长度和位置,从而量化了几种位置权重矩阵(PWM)算法的性能。我们将这些方法应用于参与细胞生长和增殖调节以及炎症、免疫和抗病毒反应(NF-κB、ISGF3、IRF1、STAT1)、肥胖和脂质代谢(PPAR、SREBP、HNF4)、类固醇生成(SF-1)和细胞周期(E2F)基因表达调节的生物医学重要TFBS。我们还使用一种名为SiteGA的方法获得了额外的特异性,该方法使用具有局部定位二核苷酸(LPD)频率判别函数的遗传算法(GA),考虑TFBS核心和侧翼区域内的结构相互作用。为确保我们方法的更高可信度,我们应用重采样刀切法和自展检验进行比较,结果表明,优化的PWM和SiteGA显示出相似的识别性能。然后我们将SiteGA和优化的PWM(单独和一起)应用于真核生物启动子数据库(EPD)中的序列。现在可以使用网络工具SiteGA将所得的SiteGA识别模型用于搜索序列中的结合位点。对SiteGA模型揭示的近距离和远距离LPD之间的依赖性分析表明,最显著的相关性存在于近距离LPD之间,并且通常位于核心(足迹)区域。大量不太显著的相关性主要存在于远距离LPD之间,其跨越核心和侧翼区域。当一起应用SiteGA和优化的PWM模型时,这至少在更高严格度下大幅减少了假阳性。
基于此分析,SiteGA即使对于优化的PWM也增加了显著的特异性,可考虑用于大规模基因组分析。它增加了可用于TFBS预测的技术范围,并且EPD分析产生了一份似乎受上述转录因子调节的基因列表。