Huang Weichun, Umbach David M, Ohler Uwe, Li Leping
Bioinformatics Research Center, North Carolina State University, Raleigh, NC 27606, USA.
BMC Bioinformatics. 2006 Jun 2;7:279. doi: 10.1186/1471-2105-7-279.
Identifying functional elements, such as transcriptional factor binding sites, is a fundamental step in reconstructing gene regulatory networks and remains a challenging issue, largely due to limited availability of training samples.
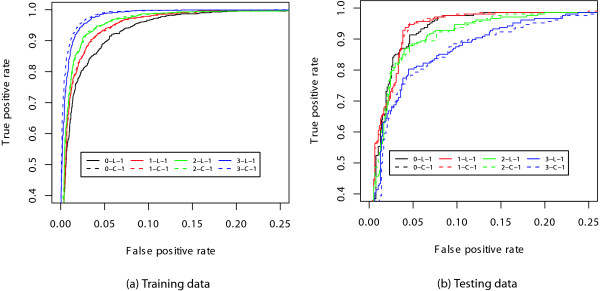
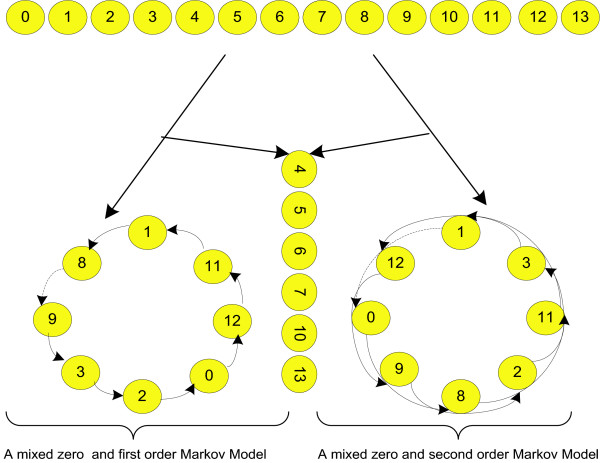
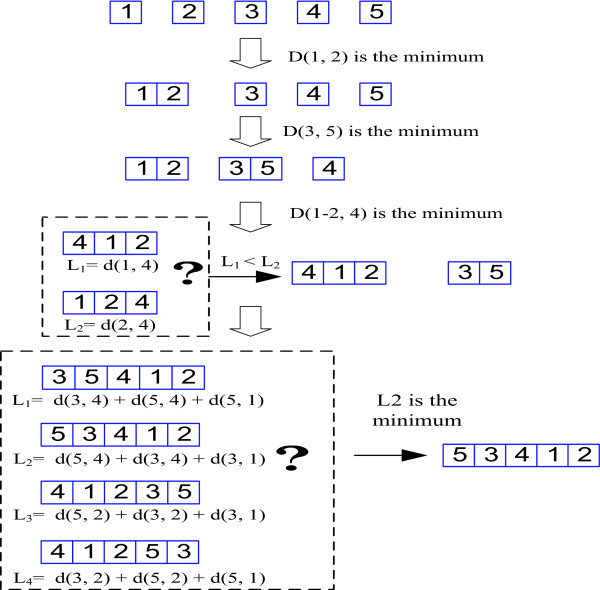
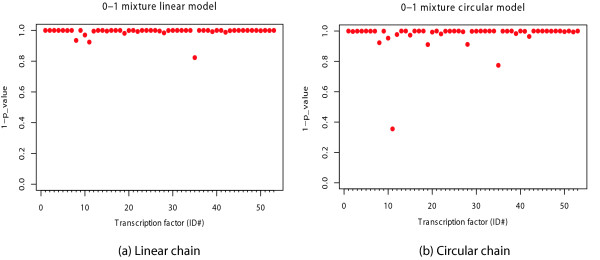
We introduce a novel and flexible model, the Optimized Mixture Markov model (OMiMa), and related methods to allow adjustment of model complexity for different motifs. In comparison with other leading methods, OMiMa can incorporate more than the NNSplice's pairwise dependencies; OMiMa avoids model over-fitting better than the Permuted Variable Length Markov Model (PVLMM); and OMiMa requires smaller training samples than the Maximum Entropy Model (MEM). Testing on both simulated and actual data (regulatory cis-elements and splice sites), we found OMiMa's performance superior to the other leading methods in terms of prediction accuracy, required size of training data or computational time. Our OMiMa system, to our knowledge, is the only motif finding tool that incorporates automatic selection of the best model. OMiMa is freely available at 1.
Our optimized mixture of Markov models represents an alternative to the existing methods for modeling dependent structures within a biological motif. Our model is conceptually simple and effective, and can improve prediction accuracy and/or computational speed over other leading methods.
识别功能元件,如转录因子结合位点,是重建基因调控网络的基本步骤,并且仍然是一个具有挑战性的问题,这主要是由于训练样本的可用性有限。
我们引入了一种新颖且灵活的模型,即优化混合马尔可夫模型(OMiMa)以及相关方法,以允许针对不同基序调整模型复杂度。与其他领先方法相比,OMiMa能够纳入比NNSplice更多的成对依赖性;OMiMa比置换可变长度马尔可夫模型(PVLMM)能更好地避免模型过拟合;并且OMiMa比最大熵模型(MEM)需要的训练样本更少。在模拟数据和实际数据(调控顺式元件和剪接位点)上进行测试时,我们发现OMiMa在预测准确性、所需训练数据大小或计算时间方面的性能优于其他领先方法。据我们所知,我们的OMiMa系统是唯一包含自动选择最佳模型的基序发现工具。OMiMa可在1免费获取。
我们优化的马尔可夫模型混合体是用于对生物基序内的依赖结构进行建模的现有方法的一种替代方案。我们的模型在概念上简单且有效,并且与其他领先方法相比能够提高预测准确性和/或计算速度。