Habeck Michael, Nilges Michael, Rieping Wolfgang
Department of Protein Evolution, Max-Planck-Institute for Developmental Biology, Spemannstr. 35, 72076, Tubingen, Germany.
J Biomol NMR. 2008 Feb;40(2):135-44. doi: 10.1007/s10858-007-9215-1. Epub 2007 Dec 20.
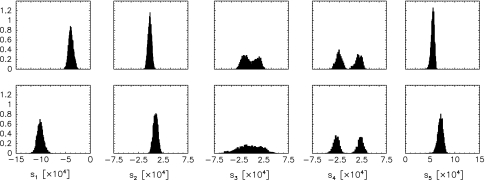
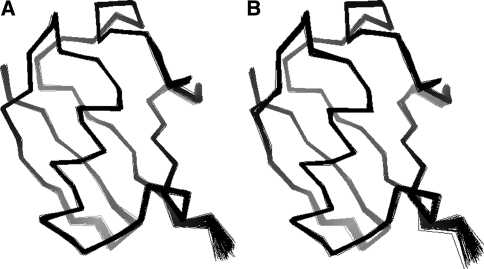
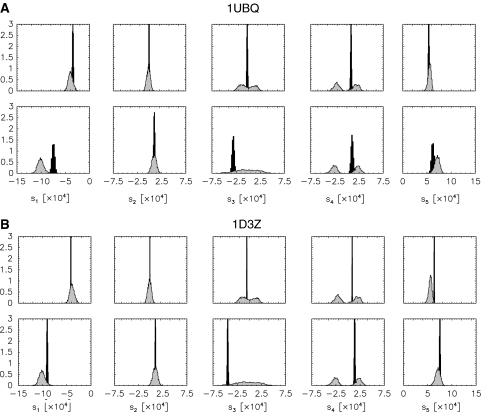
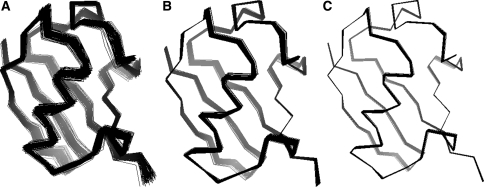
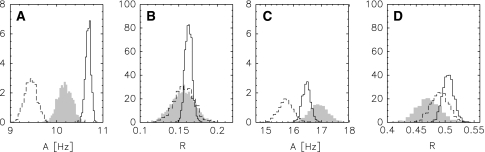
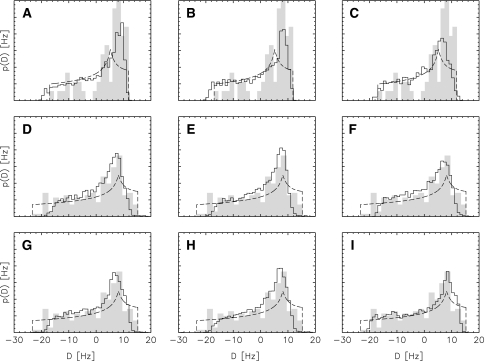
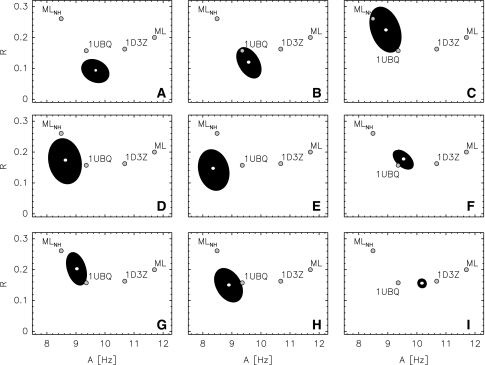
Residual dipolar couplings provide complementary information to the nuclear Overhauser effect measurements that are traditionally used in biomolecular structure determination by NMR. In a de novo structure determination, however, lack of knowledge about the degree and orientation of molecular alignment complicates the analysis of dipolar coupling data. We present a probabilistic framework for analyzing residual dipolar couplings and demonstrate that it is possible to estimate the atomic coordinates, the complete molecular alignment tensor, and the error of the couplings simultaneously. As a by-product, we also obtain estimates of the uncertainty in the coordinates and the alignment tensor. We show that our approach encompasses existing methods for determining the alignment tensor as special cases, including least squares estimation, histogram fitting, and elimination of an explicit alignment tensor in the restraint energy.
剩余偶极耦合为传统上用于通过核磁共振确定生物分子结构的核Overhauser效应测量提供了补充信息。然而,在从头进行结构确定时,缺乏关于分子排列程度和方向的知识使偶极耦合数据分析变得复杂。我们提出了一个用于分析剩余偶极耦合的概率框架,并证明可以同时估计原子坐标、完整的分子排列张量和耦合误差。作为副产品,我们还获得了坐标和排列张量不确定性的估计值。我们表明,我们的方法包含了作为特殊情况的现有确定排列张量的方法,包括最小二乘估计、直方图拟合以及在约束能量中消除显式排列张量。