Weston David J, Gunter Lee E, Rogers Alistair, Wullschleger Stan D
Environmental Sciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831-6422, USA.
BMC Syst Biol. 2008 Feb 4;2:16. doi: 10.1186/1752-0509-2-16.
One of the eminent opportunities afforded by modern genomic technologies is the potential to provide a mechanistic understanding of the processes by which genetic change translates to phenotypic variation and the resultant appearance of distinct physiological traits. Indeed much progress has been made in this area, particularly in biomedicine where functional genomic information can be used to determine the physiological state (e.g., diagnosis) and predict phenotypic outcome (e.g., patient survival). Ecology currently lacks an analogous approach where genomic information can be used to diagnose the presence of a given physiological state (e.g., stress response) and then predict likely phenotypic outcomes (e.g., stress duration and tolerance, fitness).
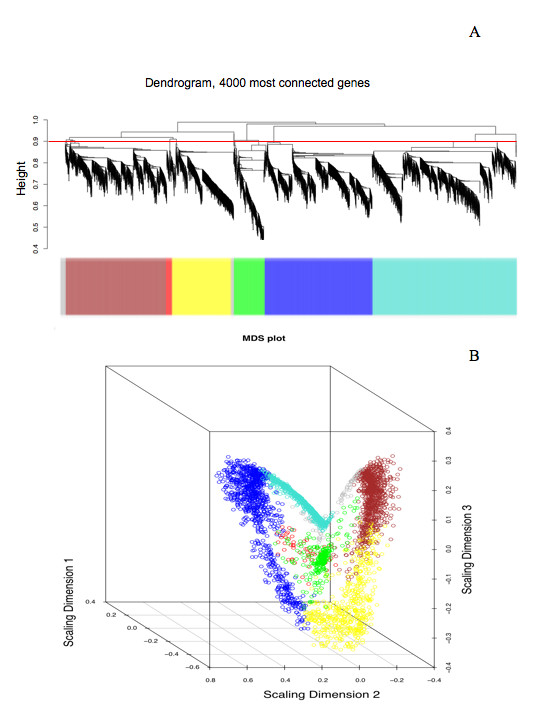
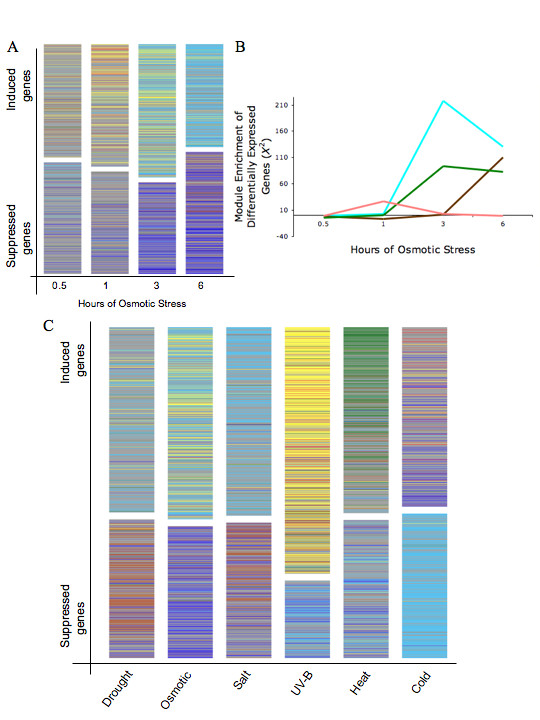
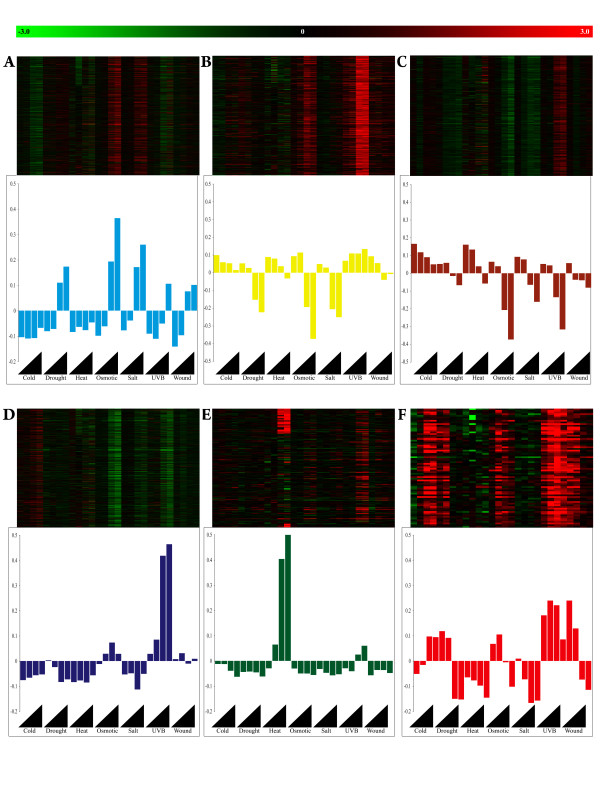
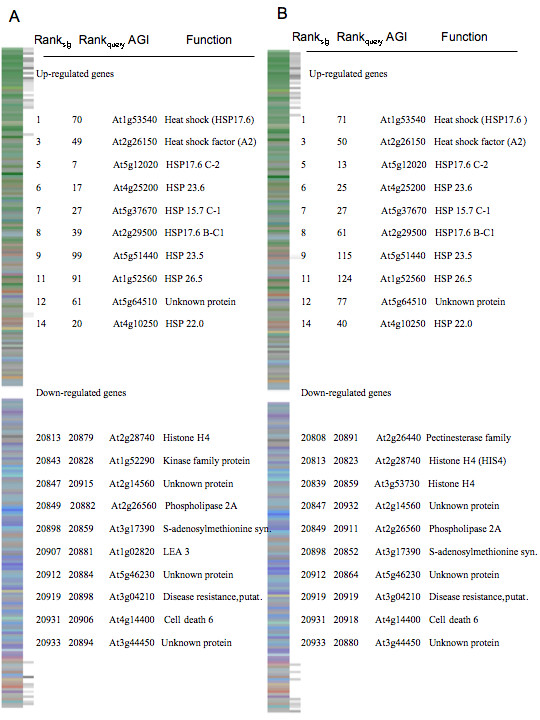
Here, we demonstrate that a compendium of genomic signatures can be used to classify the plant abiotic stress phenotype in Arabidopsis according to the architecture of the transcriptome, and then be linked with gene coexpression network analysis to determine the underlying genes governing the phenotypic response. Using this approach, we confirm the existence of known stress responsive pathways and marker genes, report a common abiotic stress responsive transcriptome and relate phenotypic classification to stress duration.
Linking genomic signatures to gene coexpression analysis provides a unique method of relating an observed plant phenotype to changes in gene expression that underlie that phenotype. Such information is critical to current and future investigations in plant biology and, in particular, to evolutionary ecology, where a mechanistic understanding of adaptive physiological responses to abiotic stress can provide researchers with a tool of great predictive value in understanding species and population level adaptation to climate change.
现代基因组技术带来的一个显著机遇是,有可能对遗传变化转化为表型变异以及由此产生不同生理特征的过程提供机制性理解。事实上,在这一领域已经取得了很大进展,特别是在生物医学领域,功能基因组信息可用于确定生理状态(如诊断)并预测表型结果(如患者存活率)。目前生态学缺乏一种类似的方法,即利用基因组信息来诊断给定生理状态(如应激反应)的存在,然后预测可能的表型结果(如应激持续时间和耐受性、适应性)。
在这里,我们证明了一组基因组特征可根据转录组结构对拟南芥中的植物非生物胁迫表型进行分类,然后与基因共表达网络分析相结合,以确定控制表型反应的潜在基因。使用这种方法,我们证实了已知的应激反应途径和标记基因的存在,报告了一个常见的非生物胁迫反应转录组,并将表型分类与胁迫持续时间联系起来。
将基因组特征与基因共表达分析联系起来,提供了一种将观察到的植物表型与构成该表型基础的基因表达变化相关联的独特方法。这些信息对于当前和未来的植物生物学研究至关重要,特别是对于进化生态学而言,其中对非生物胁迫的适应性生理反应的机制性理解可以为研究人员提供一个具有巨大预测价值的工具,以理解物种和种群水平对气候变化的适应性。