Kang Silvia S, Bloom Seth M, Norian Lyse A, Geske Michael J, Flavell Richard A, Stappenbeck Thaddeus S, Allen Paul M
Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, Missouri, United States of America.
PLoS Med. 2008 Mar 4;5(3):e41. doi: 10.1371/journal.pmed.0050041.
The constellation of human inflammatory bowel disease (IBD) includes ulcerative colitis and Crohn's disease, which both display a wide spectrum in the severity of pathology. One theory is that multiple genetic hits to the host immune system may contribute to the susceptibility and severity of IBD. However, experimental proof of this concept is still lacking. Several genetic mouse models that each recapitulate some aspects of human IBD have utilized a single gene defect to induce colitis. However, none have produced pathology clearly distinguishable as either ulcerative colitis or Crohn's disease, in part because none of them reproduce the most severe forms of disease that are observed in human patients. This lack of severe IBD models has posed a challenge for research into pathogenic mechanisms and development of new treatments. We hypothesized that multiple genetic hits to the regulatory machinery that normally inhibits immune activation in the intestine would generate more severe, reproducible pathology that would mimic either ulcerative colitis or Crohn's disease.
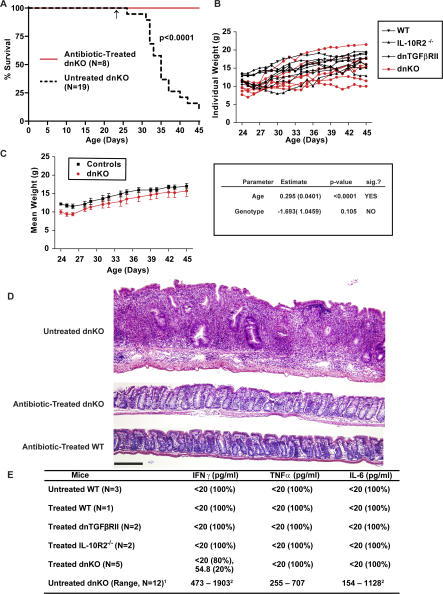
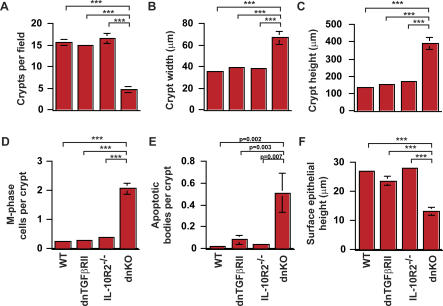
We generated a novel mouse line (dnKO) that possessed defects in both TGFbetaRII and IL-10R2 signaling. These mice rapidly and reproducibly developed a disease resembling fulminant human ulcerative colitis that was quite distinct from the much longer and more variable course of pathology observed previously in mice possessing only single defects. Pathogenesis was driven by uncontrolled production of proinflammatory cytokines resulting in large part from T cell activation. The disease process could be significantly ameliorated by administration of antibodies against IFNgamma and TNFalpha and was completely inhibited by a combination of broad-spectrum antibiotics.
Here, we develop to our knowledge the first mouse model of fulminant ulcerative colitis by combining multiple genetic hits in immune regulation and demonstrate that the resulting disease is sensitive to both anticytokine therapy and broad-spectrum antibiotics. These findings indicated the IL-10 and TGFbeta pathways synergize to inhibit microbially induced production of proinflammatory cytokines, including IFNgamma and TNFalpha, which are known to play a role in the pathogenesis of human ulcerative colitis. Our findings also provide evidence that broad-spectrum antibiotics may have an application in the treatment of patients with ulcerative colitis. This model system will be useful in the future to explore the microbial factors that induce immune activation and characterize how these interactions produce disease.
人类炎症性肠病(IBD)包括溃疡性结肠炎和克罗恩病,二者在病理严重程度上均表现出广泛的谱系。一种理论认为,宿主免疫系统受到多次基因打击可能导致IBD的易感性和严重程度。然而,这一概念的实验证据仍然缺乏。几种分别概括人类IBD某些方面的基因小鼠模型利用单一基因缺陷来诱发结肠炎。然而,没有一种模型产生的病理表现能明确区分为溃疡性结肠炎或克罗恩病,部分原因是它们都没有重现人类患者中观察到的最严重的疾病形式。缺乏严重的IBD模型对致病机制的研究和新疗法的开发构成了挑战。我们假设,对正常情况下抑制肠道免疫激活的调节机制进行多次基因打击会产生更严重、可重复的病理表现,从而模拟溃疡性结肠炎或克罗恩病。
我们培育出一种新型小鼠品系(dnKO),其在转化生长因子β受体II(TGFbetaRII)和白细胞介素10受体2(IL-10R2)信号传导方面均存在缺陷。这些小鼠迅速且可重复地发展出一种类似于暴发性人类溃疡性结肠炎的疾病,这与之前在仅具有单一缺陷的小鼠中观察到的病程长得多且更具变异性的病理情况截然不同。发病机制是由促炎细胞因子的不受控制产生所驱动,这在很大程度上源于T细胞激活。通过给予抗干扰素γ(IFNgamma)和肿瘤坏死因子α(TNFalpha)的抗体,疾病进程可得到显著改善,而广谱抗生素的联合使用可完全抑制该疾病进程。
在此,据我们所知,我们通过在免疫调节中结合多次基因打击,开发出了首个暴发性溃疡性结肠炎小鼠模型,并证明所产生的疾病对抗细胞因子疗法和广谱抗生素均敏感。这些发现表明,白细胞介素10(IL-10)和转化生长因子β(TGFbeta)途径协同抑制微生物诱导的促炎细胞因子的产生,包括已知在人类溃疡性结肠炎发病机制中起作用的干扰素γ和肿瘤坏死因子α。我们的发现还提供了证据表明广谱抗生素可能在溃疡性结肠炎患者的治疗中具有应用价值。这个模型系统未来将有助于探索诱导免疫激活的微生物因素,并表征这些相互作用如何引发疾病。