Meltzer Eric B, Noble Paul W
Department of Medicine, Division of Pulmonary, Allergy and Critical Care, Duke University Medical Center, Durham, North Carolina 27710, USA.
Orphanet J Rare Dis. 2008 Mar 26;3:8. doi: 10.1186/1750-1172-3-8.
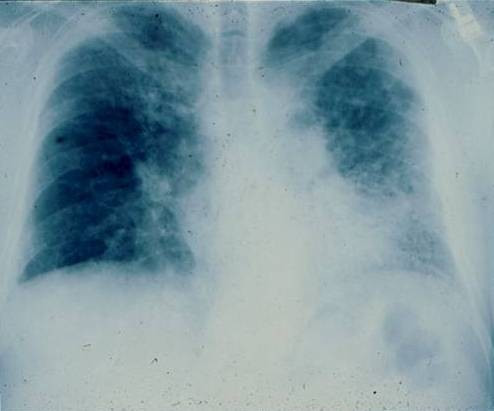
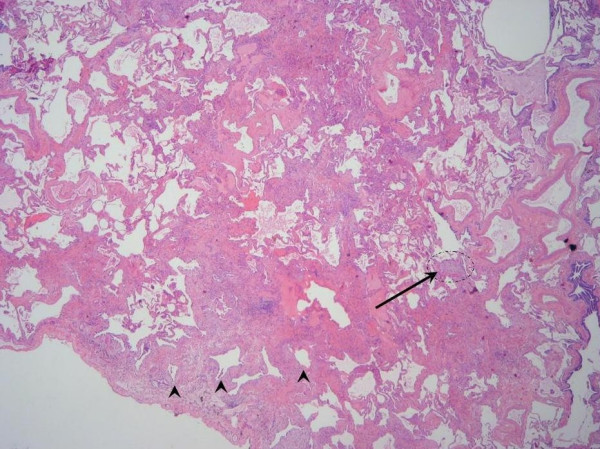
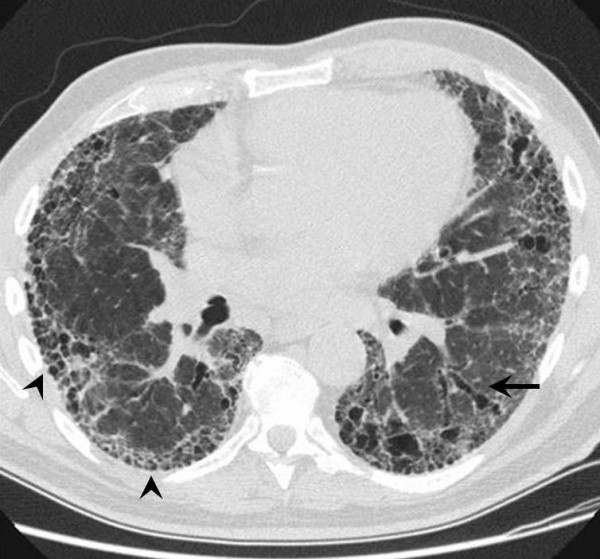
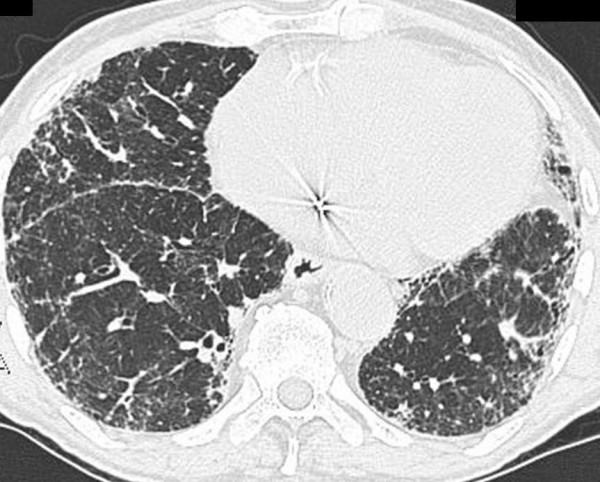
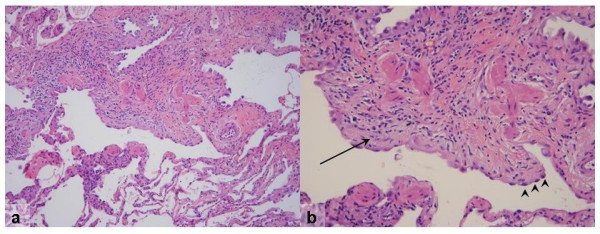
Idiopathic pulmonary fibrosis (IPF) is a non-neoplastic pulmonary disease that is characterized by the formation of scar tissue within the lungs in the absence of any known provocation. IPF is a rare disease which affects approximately 5 million persons worldwide. The prevalence is estimated to be slightly greater in men (20.2/100,000) than in women (13.2/100,000). The mean age at presentation is 66 years. IPF initially manifests with symptoms of exercise-induced breathless and dry coughing. Auscultation of the lungs reveals early inspiratory crackles, predominantly located in the lower posterior lung zones upon physical exam. Clubbing is found in approximately 50% of IPF patients. Cor pulmonale develops in association with end-stage disease. In that case, classic signs of right heart failure may be present. Etiology remains incompletely understood. Some environmental factors may be associated with IPF (cigarette smoking, exposure to silica and livestock). IPF is recognized on high-resolution computed tomography by peripheral, subpleural lower lobe reticular opacities in association with subpleural honeycomb changes. IPF is associated with a pathological lesion known as usual interstitial pneumonia (UIP). The UIP pattern consists of normal lung alternating with patches of dense fibrosis, taking the form of collagen sheets. The diagnosis of IPF requires correlation of the clinical setting with radiographic images and a lung biopsy. In the absence of lung biopsy, the diagnosis of IPF can be made by defined clinical criteria that were published in guidelines endorsed by several professional societies. Differential diagnosis includes other idiopathic interstitial pneumonia, connective tissue diseases (systemic sclerosis, polymyositis, rheumatoid arthritis), forme fruste of autoimmune disorders, chronic hypersensitivity pneumonitis and other environmental (sometimes occupational) exposures. IPF is typically progressive and leads to significant disability. The median survival is 2 to 5 years from the time of diagnosis. Medical therapy is ineffective in the treatment of IPF. New molecular therapeutic targets have been identified and several clinical trials are investigating the efficacy of novel medication. Meanwhile, pulmonary transplantation remains a viable option for patients with IPF. It is expected that, during the next decade, considerable progress will be made toward the understanding and treatment of this devastating illness.
特发性肺纤维化(IPF)是一种非肿瘤性肺部疾病,其特征是在没有任何已知诱因的情况下,肺部形成瘢痕组织。IPF是一种罕见疾病,全球约有500万人受其影响。据估计,男性患病率(20.2/10万)略高于女性(13.2/10万)。发病时的平均年龄为66岁。IPF最初表现为运动性呼吸困难和干咳症状。肺部听诊可发现早期吸气性啰音,体格检查时主要位于肺下后区。约50%的IPF患者会出现杵状指。肺心病与终末期疾病相关。在这种情况下,可能会出现右心衰竭的典型体征。病因仍未完全明确。一些环境因素可能与IPF有关(吸烟、接触二氧化硅和家畜)。在高分辨率计算机断层扫描上,IPF表现为外周、胸膜下肺下叶网状阴影,并伴有胸膜下蜂窝状改变。IPF与一种称为普通型间质性肺炎(UIP)的病理病变相关。UIP模式表现为正常肺组织与密集纤维化区域交替出现,呈胶原纤维束形式。IPF的诊断需要结合临床情况、影像学图像和肺活检结果。在没有肺活检的情况下,可根据多个专业学会认可的指南中规定的临床标准进行IPF诊断。鉴别诊断包括其他特发性间质性肺炎、结缔组织病(系统性硬化症、多发性肌炎、类风湿关节炎)、自身免疫性疾病的顿挫型、慢性过敏性肺炎以及其他环境(有时是职业性)暴露。IPF通常呈进行性发展,会导致严重残疾。从诊断时起,中位生存期为2至5年。药物治疗对IPF无效。已确定了新的分子治疗靶点,多项临床试验正在研究新型药物的疗效。同时,肺移植仍是IPF患者的可行选择。预计在未来十年,在对这种毁灭性疾病的认识和治疗方面将取得重大进展。