Liu Ying, Eyal Eran, Bahar Ivet
Department of Computational Biology, School of Medicine, University of Pittsburgh, PA 15232, USA.
Bioinformatics. 2008 May 15;24(10):1243-50. doi: 10.1093/bioinformatics/btn110. Epub 2008 Mar 28.
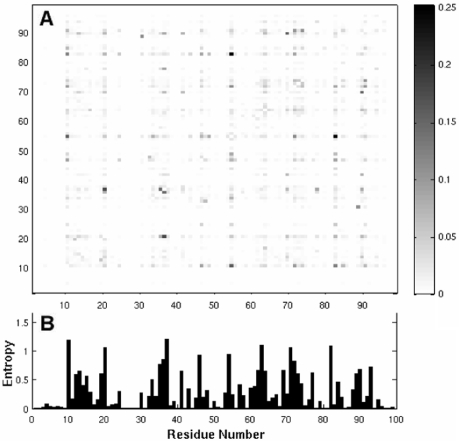
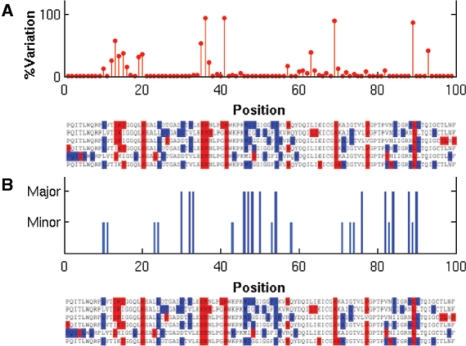
The ability of human immunodeficiency virus-1 (HIV-1) protease to develop mutations that confer multi-drug resistance (MDR) has been a major obstacle in designing rational therapies against HIV. Resistance is usually imparted by a cooperative mechanism that can be elucidated by a covariance analysis of sequence data. Identification of such correlated substitutions of amino acids may be obscured by evolutionary noise.
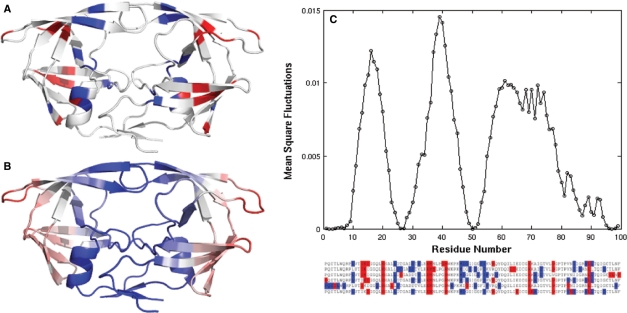
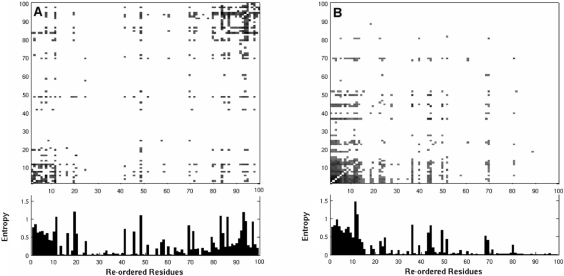
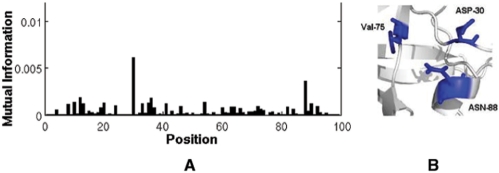
HIV-1 protease sequences from patients subjected to different specific treatments (set 1), and from untreated patients (set 2) were subjected to sequence covariance analysis by evaluating the mutual information (MI) between all residue pairs. Spectral clustering of the resulting covariance matrices disclosed two distinctive clusters of correlated residues: the first, observed in set 1 but absent in set 2, contained residues involved in MDR acquisition; and the second, included those residues differentiated in the various HIV-1 protease subtypes, shortly referred to as the phylogenetic cluster. The MDR cluster occupies sites close to the central symmetry axis of the enzyme, which overlap with the global hinge region identified from coarse-grained normal-mode analysis of the enzyme structure. The phylogenetic cluster, on the other hand, occupies solvent-exposed and highly mobile regions. This study demonstrates (i) the possibility of distinguishing between the correlated substitutions resulting from neutral mutations and those induced by MDR upon appropriate clustering analysis of sequence covariance data and (ii) a connection between global dynamics and functional substitution of amino acids.
人类免疫缺陷病毒1型(HIV-1)蛋白酶产生赋予多药耐药性(MDR)突变的能力,一直是设计针对HIV的合理疗法的主要障碍。耐药性通常由一种协同机制赋予,这种机制可通过序列数据的协方差分析来阐明。氨基酸的这种相关替换的识别可能会被进化噪声所掩盖。
对接受不同特定治疗的患者(第1组)和未接受治疗的患者(第2组)的HIV-1蛋白酶序列进行了序列协方差分析,通过评估所有残基对之间的互信息(MI)。对所得协方差矩阵进行谱聚类,揭示了两个不同的相关残基簇:第一个在第1组中观察到但在第2组中不存在,包含参与获得MDR的残基;第二个包括在各种HIV-1蛋白酶亚型中有所差异的那些残基,简称为系统发育簇。MDR簇占据靠近酶中心对称轴的位点,这些位点与从酶结构的粗粒度正常模式分析中确定的全局铰链区域重叠。另一方面,系统发育簇占据溶剂暴露且高度可变的区域。这项研究表明:(i)在对序列协方差数据进行适当的聚类分析后,有可能区分由中性突变导致的相关替换和由MDR诱导的相关替换;(ii)全局动力学与氨基酸功能替换之间的联系。