Firth Andrew E, Patrick Wayne M
BioSciences Institute, University College Cork, Cork, Ireland.
Nucleic Acids Res. 2008 Jul 1;36(Web Server issue):W281-5. doi: 10.1093/nar/gkn226. Epub 2008 Apr 28.
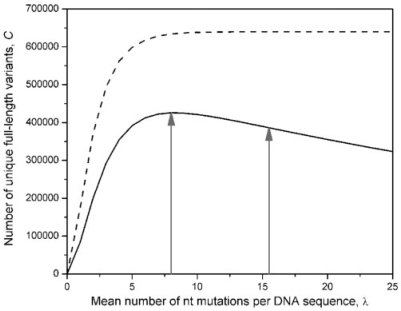
There are many methods for introducing random mutations into nucleic acid sequences. Previously, we described a suite of programmes for estimating the completeness and diversity of randomized DNA libraries generated by a number of these protocols. Our programmes suggested some empirical guidelines for library design; however, no information was provided regarding library diversity at the protein (rather than DNA) level. We have now updated our web server, enabling analysis of translated libraries constructed by site-saturation mutagenesis and error-prone PCR (epPCR). We introduce GLUE-Including Translation (GLUE-IT), which finds the expected amino acid completeness of libraries in which up to six codons have been independently varied (according to any user-specified randomization scheme). We provide two tools for assisting with experimental design: CodonCalculator, for assessing amino acids corresponding to given randomized codons; and AA-Calculator, for finding degenerate codons that encode user-specified sets of amino acids. We also present PEDEL-AA, which calculates amino acid statistics for libraries generated by epPCR. Input includes the parent sequence, overall mutation rate, library size, indel rates and a nucleotide mutation matrix. Output includes amino acid completeness and diversity statistics, and the number and length distribution of sequences truncated by premature termination codons. The web interfaces are available at http://guinevere.otago.ac.nz/stats.html.
将随机突变引入核酸序列的方法有很多。此前,我们描述了一套程序,用于评估由许多此类方案生成的随机DNA文库的完整性和多样性。我们的程序提出了一些文库设计的经验指导原则;然而,未提供有关蛋白质(而非DNA)水平文库多样性的信息。我们现已更新了我们的网络服务器,能够分析通过位点饱和诱变和易错PCR(epPCR)构建的翻译文库。我们引入了包含翻译的GLUE(GLUE-IT),它能找到在多达六个密码子已独立变化(根据任何用户指定的随机化方案)的文库中预期的氨基酸完整性。我们提供了两个辅助实验设计的工具:CodonCalculator,用于评估与给定随机密码子对应的氨基酸;以及AA-Calculator,用于找到编码用户指定氨基酸集的简并密码子。我们还展示了PEDEL-AA,它计算由epPCR生成的文库的氨基酸统计数据。输入包括亲本序列、总体突变率、文库大小、插入缺失率和核苷酸突变矩阵。输出包括氨基酸完整性和多样性统计数据,以及被提前终止密码子截断的序列的数量和长度分布。网络界面可在http://guinevere.otago.ac.nz/stats.html获取。