Cardona Gabriel, Llabrés Mercè, Rosselló Francesc, Valiente Gabriel
Department of Mathematics and Computer Science, University of the Balearic Islands, E-07122 Palma de Mallorca, Spain.
Bioinformatics. 2008 Jul 1;24(13):1481-8. doi: 10.1093/bioinformatics/btn231. Epub 2008 May 12.
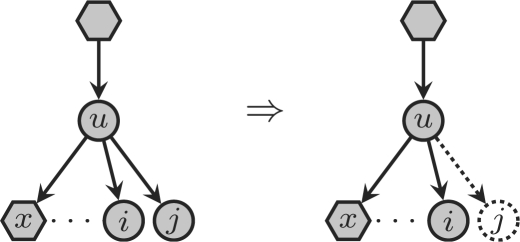
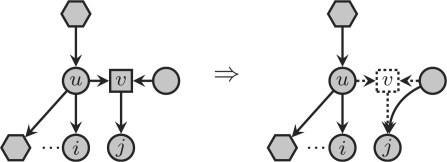
The presence of reticulate evolutionary events in phylogenies turn phylogenetic trees into phylogenetic networks. These events imply in particular that there may exist multiple evolutionary paths from a non-extant species to an extant one, and this multiplicity makes the comparison of phylogenetic networks much more difficult than the comparison of phylogenetic trees. In fact, all attempts to define a sound distance measure on the class of all phylogenetic networks have failed so far. Thus, the only practical solutions have been either the use of rough estimates of similarity (based on comparison of the trees embedded in the networks), or narrowing the class of phylogenetic networks to a certain class where such a distance is known and can be efficiently computed. The first approach has the problem that one may identify two networks as equivalent, when they are not; the second one has the drawback that there may not exist algorithms to reconstruct such networks from biological sequences.
We present in this article a distance measure on the class of semi-binary tree-sibling time consistent phylogenetic networks, which generalize tree-child time consistent phylogenetic networks, and thus also galled-trees. The practical interest of this distance measure is 2-fold: it can be computed in polynomial time by means of simple algorithms, and there also exist polynomial-time algorithms for reconstructing networks of this class from DNA sequence data.
The Perl package Bio::PhyloNetwork, included in the BioPerl bundle, implements many algorithms on phylogenetic networks, including the computation of the distance presented in this article.
Some counterexamples, proofs of the results not included in this article, and some computational experiments are available at Bioinformatics online.
系统发育树中网状进化事件的存在将系统发育树转变为系统发育网络。这些事件尤其意味着从一个已灭绝物种到一个现存物种可能存在多条进化路径,这种多样性使得系统发育网络的比较比系统发育树的比较困难得多。事实上,到目前为止,所有试图在所有系统发育网络类上定义合理距离度量的尝试都失败了。因此,唯一实际的解决方案要么是使用相似性的粗略估计(基于网络中嵌入树的比较),要么将系统发育网络类缩小到已知并能有效计算这种距离的特定类。第一种方法的问题是,可能会将两个并不等价的网络识别为等价;第二种方法的缺点是可能不存在从生物序列重建此类网络的算法。
我们在本文中提出了一种对半二叉树-兄弟时间一致系统发育网络类的距离度量,它推广了树子时间一致系统发育网络,因此也推广了有结树。这种距离度量的实际意义有两方面:它可以通过简单算法在多项式时间内计算,并且也存在从DNA序列数据重建此类网络的多项式时间算法。
包含在BioPerl包中的Perl包Bio::PhyloNetwork实现了许多关于系统发育网络的算法,包括本文中提出的距离计算。
一些反例、本文未包含的结果证明以及一些计算实验可在《生物信息学》在线获取。