Bush William S, Edwards Todd L, Dudek Scott M, McKinney Brett A, Ritchie Marylyn D
Center for Human Genetics Research, Department of Molecular Physiology and Biophysics, Vanderbilt University Medical Center, Nashville, Tennessee, USA.
BMC Bioinformatics. 2008 May 16;9:238. doi: 10.1186/1471-2105-9-238.
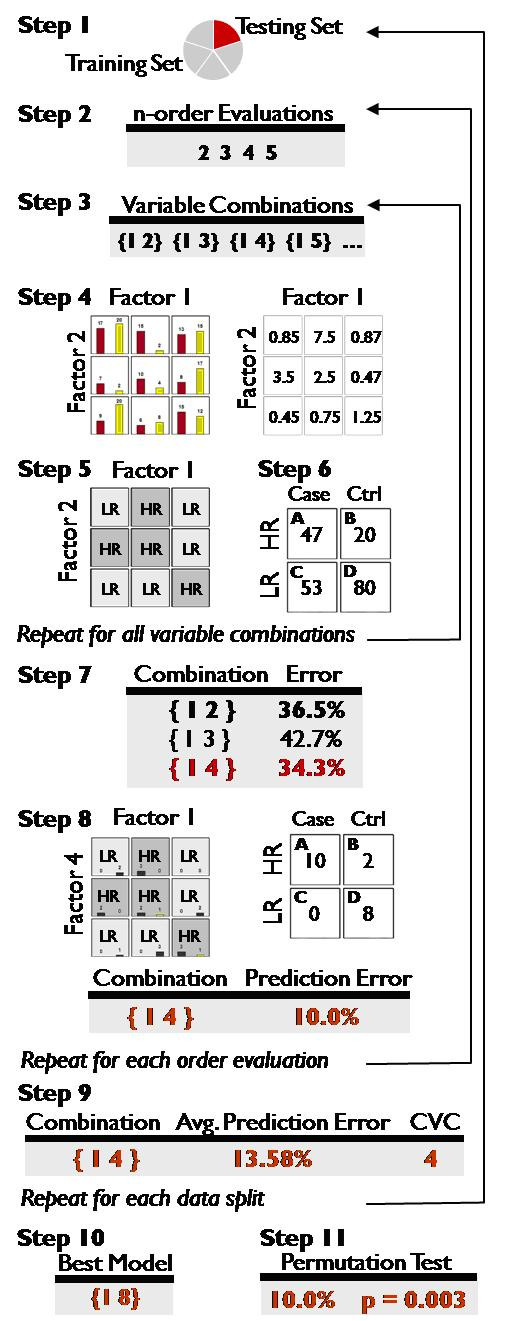
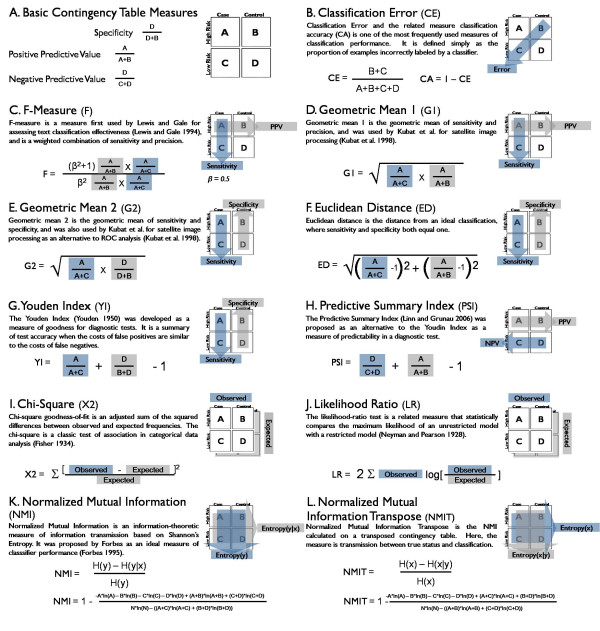
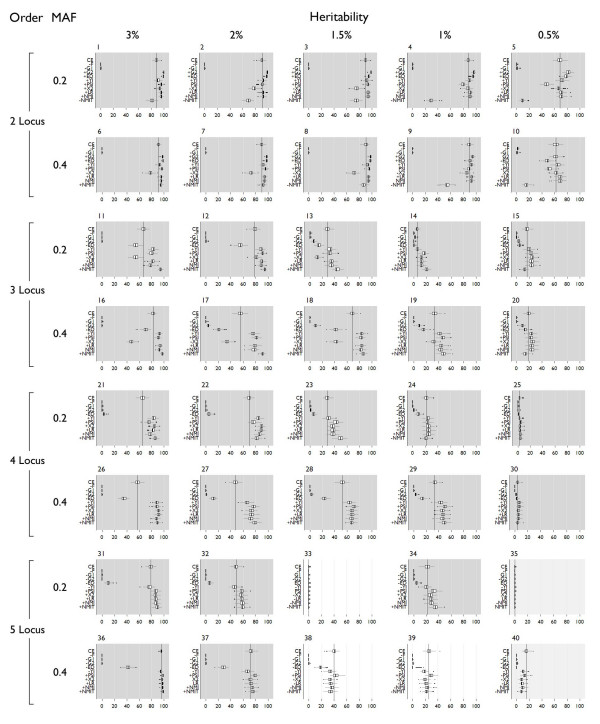
Multifactor Dimensionality Reduction (MDR) has been introduced previously as a non-parametric statistical method for detecting gene-gene interactions. MDR performs a dimensional reduction by assigning multi-locus genotypes to either high- or low-risk groups and measuring the percentage of cases and controls incorrectly labelled by this classification - the classification error. The combination of variables that produces the lowest classification error is selected as the best or most fit model. The correctly and incorrectly labelled cases and controls can be expressed as a two-way contingency table. We sought to improve the ability of MDR to detect gene-gene interactions by replacing classification error with a different measure to score model quality.
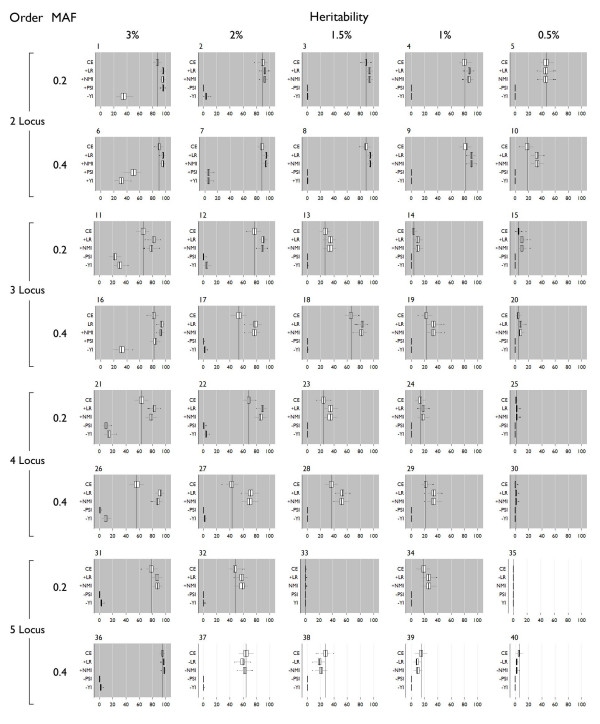
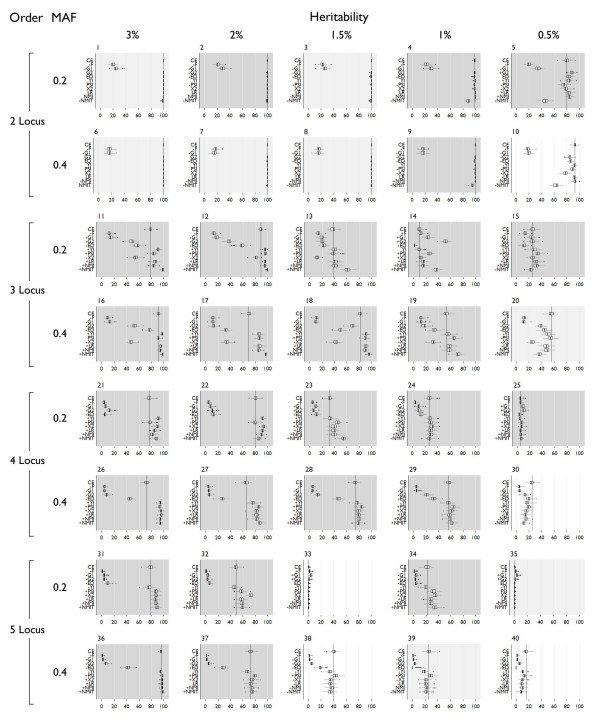
In this study, we compare the detection and power of MDR using a variety of measures for two-way contingency table analysis. We simulated 40 genetic models, varying the number of disease loci in the model (2 - 5), allele frequencies of the disease loci (.2/.8 or .4/.6) and the broad-sense heritability of the model (.05 - .3). Overall, detection using NMI was 65.36% across all models, and specific detection was 59.4% versus detection using classification error at 62% and specific detection was 52.2%.
Of the 10 measures evaluated, the likelihood ratio and normalized mutual information (NMI) are measures that consistently improve the detection and power of MDR in simulated data over using classification error. These measures also reduce the inclusion of spurious variables in a multi-locus model. Thus, MDR, which has already been demonstrated as a powerful tool for detecting gene-gene interactions, can be improved with the use of alternative fitness functions.
多因素降维法(MDR)此前已作为一种用于检测基因-基因相互作用的非参数统计方法被引入。MDR通过将多位点基因型分为高风险或低风险组并测量被此分类错误标记的病例和对照的百分比(即分类错误)来进行降维。产生最低分类错误的变量组合被选为最佳或最适合的模型。正确和错误标记的病例与对照可以表示为一个双向列联表。我们试图通过用一种不同的模型质量评分方法取代分类错误来提高MDR检测基因-基因相互作用的能力。
在本研究中,我们比较了使用多种方法进行双向列联表分析时MDR的检测能力和效能。我们模拟了40种遗传模型,改变模型中疾病位点的数量(2 - 5个)、疾病位点的等位基因频率(0.2/0.8或0.4/0.6)以及模型的广义遗传率(0.05 - 0.3)。总体而言,在所有模型中使用归一化互信息(NMI)的检测率为65.36%,特异性检测率为59.4%,而使用分类错误时的检测率为62%,特异性检测率为52.2%。
在所评估的10种方法中,似然比和归一化互信息(NMI)相比于使用分类错误能持续提高MDR在模拟数据中的检测能力和效能。这些方法还减少了多位点模型中虚假变量的纳入。因此,已经被证明是检测基因-基因相互作用的强大工具的MDR,可通过使用替代的适应度函数得到改进。