Gesell Tanja, Washietl Stefan
Center for Integrative Bioinformatics Vienna, Max F. Perutz Laboratories, Dr. Bohr-Gasse 9, A-1030 Vienna, Austria.
BMC Bioinformatics. 2008 May 27;9:248. doi: 10.1186/1471-2105-9-248.
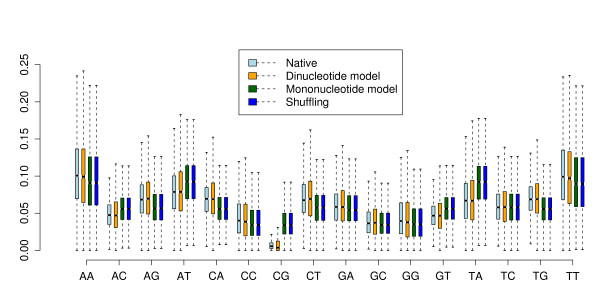
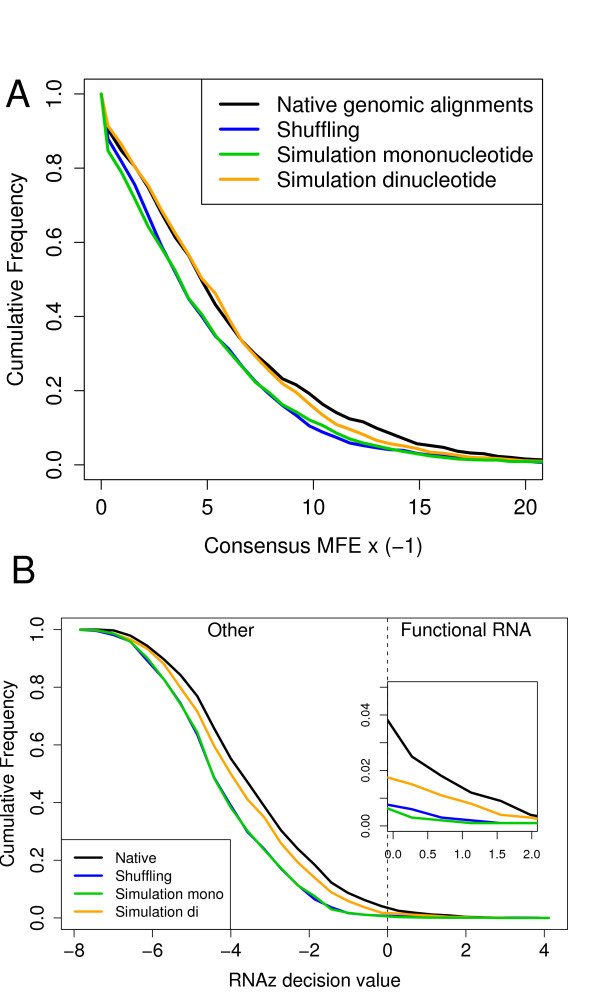
Comparative prediction of RNA structures can be used to identify functional noncoding RNAs in genomic screens. It was shown recently by Babak et al. [BMC Bioinformatics. 8:33] that RNA gene prediction programs can be biased by the genomic dinucleotide content, in particular those programs using a thermodynamic folding model including stacking energies. As a consequence, there is need for dinucleotide-preserving control strategies to assess the significance of such predictions. While there have been randomization algorithms for single sequences for many years, the problem has remained challenging for multiple alignments and there is currently no algorithm available.
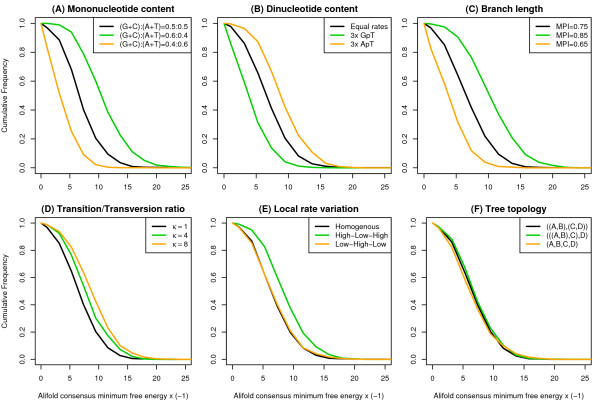
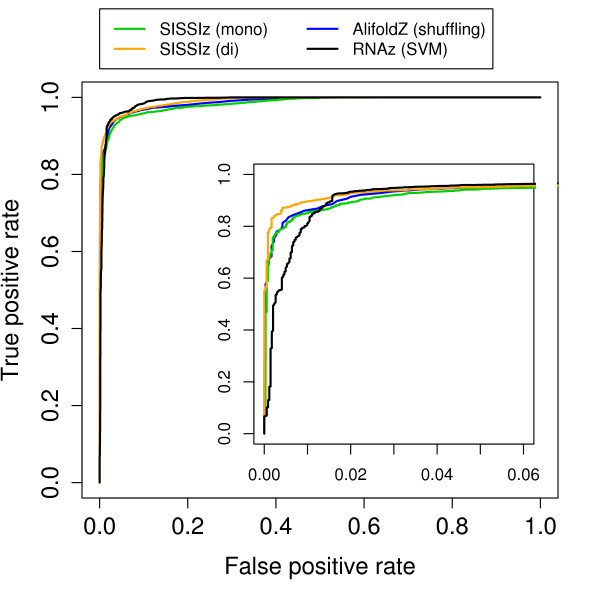
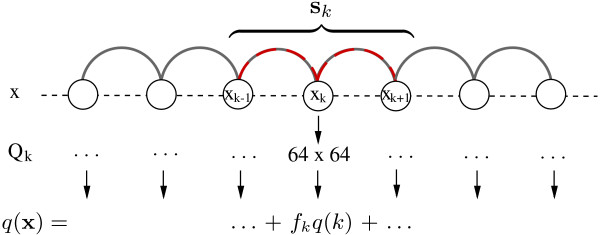
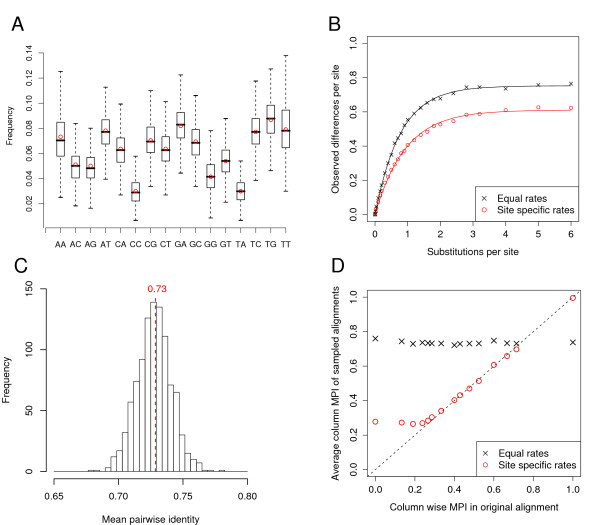
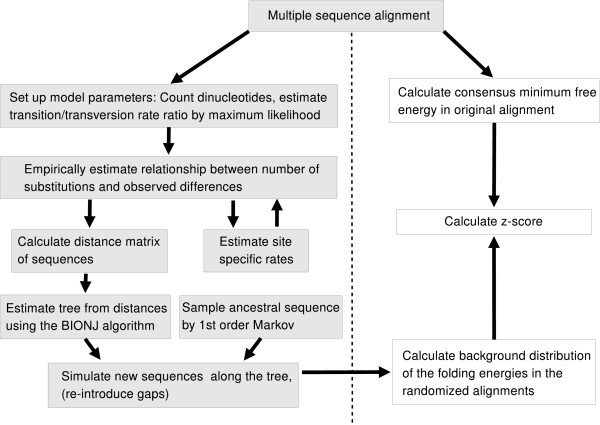
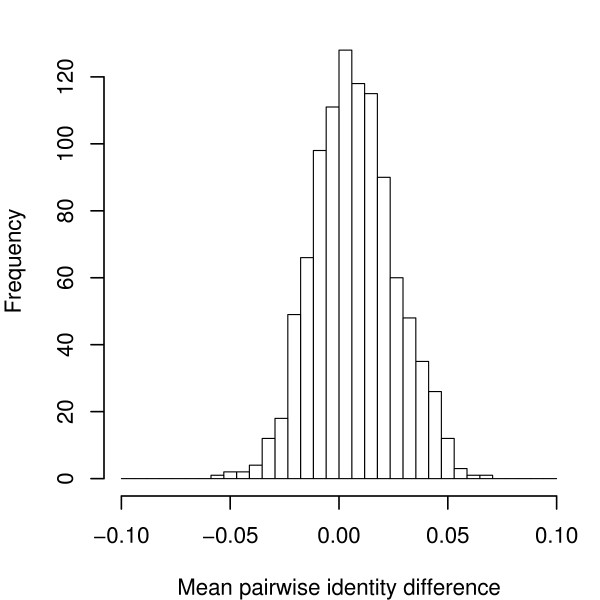
We present a program called SISSIz that simulates multiple alignments of a given average dinucleotide content. Meeting additional requirements of an accurate null model, the randomized alignments are on average of the same sequence diversity and preserve local conservation and gap patterns. We make use of a phylogenetic substitution model that includes overlapping dependencies and site-specific rates. Using fast heuristics and a distance based approach, a tree is estimated under this model which is used to guide the simulations. The new algorithm is tested on vertebrate genomic alignments and the effect on RNA structure predictions is studied. In addition, we directly combined the new null model with the RNAalifold consensus folding algorithm giving a new variant of a thermodynamic structure based RNA gene finding program that is not biased by the dinucleotide content.
SISSIz implements an efficient algorithm to randomize multiple alignments preserving dinucleotide content. It can be used to get more accurate estimates of false positive rates of existing programs, to produce negative controls for the training of machine learning based programs, or as standalone RNA gene finding program. Other applications in comparative genomics that require randomization of multiple alignments can be considered.
SISSIz is available as open source C code that can be compiled for every major platform and downloaded here: http://sourceforge.net/projects/sissiz.
RNA结构的比较预测可用于在基因组筛选中识别功能性非编码RNA。Babak等人[《BMC生物信息学》。8:33]最近表明,RNA基因预测程序可能会受到基因组二核苷酸含量的影响,特别是那些使用包括堆积能量的热力学折叠模型的程序。因此,需要采用保留二核苷酸的控制策略来评估此类预测的重要性。虽然多年来已经有针对单序列的随机化算法,但对于多序列比对来说,这个问题仍然具有挑战性,目前还没有可用的算法。
我们提出了一个名为SISSIz的程序,它可以模拟具有给定平均二核苷酸含量的多序列比对。满足准确零模型的其他要求后,随机化的比对平均具有相同的序列多样性,并保留局部保守性和空位模式。我们使用了一种系统发育替代模型,该模型包括重叠依赖性和位点特异性速率。通过快速启发式算法和基于距离的方法,在此模型下估计一棵树,并用它来指导模拟。新算法在脊椎动物基因组比对上进行了测试,并研究了其对RNA结构预测的影响。此外,我们直接将新的零模型与RNAalifold一致性折叠算法相结合,得到了一种基于热力学结构的RNA基因发现程序的新变体,该程序不受二核苷酸含量的影响。
SISSIz实现了一种有效的算法,用于随机化保留二核苷酸含量的多序列比对。它可用于更准确地估计现有程序的假阳性率,为基于机器学习的程序训练生成阴性对照,或作为独立的RNA基因发现程序。也可以考虑在比较基因组学中其他需要随机化多序列比对的应用。
SISSIz以开源C代码形式提供,可以为每个主要平台进行编译,并可在此处下载:http://sourceforge.net/projects/sissiz 。