Hannes F D, Sharp A J, Mefford H C, de Ravel T, Ruivenkamp C A, Breuning M H, Fryns J-P, Devriendt K, Van Buggenhout G, Vogels A, Stewart H, Hennekam R C, Cooper G M, Regan R, Knight S J L, Eichler E E, Vermeesch J R
Center for Human Genetics, Herestraat 49, 3000 Leuven, Belgium.
J Med Genet. 2009 Apr;46(4):223-32. doi: 10.1136/jmg.2007.055202. Epub 2008 Jun 11.
Genomic disorders are often caused by non-allelic homologous recombination between segmental duplications. Chromosome 16 is especially rich in a chromosome-specific low copy repeat, termed LCR16.
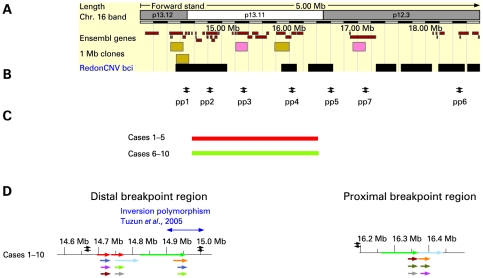
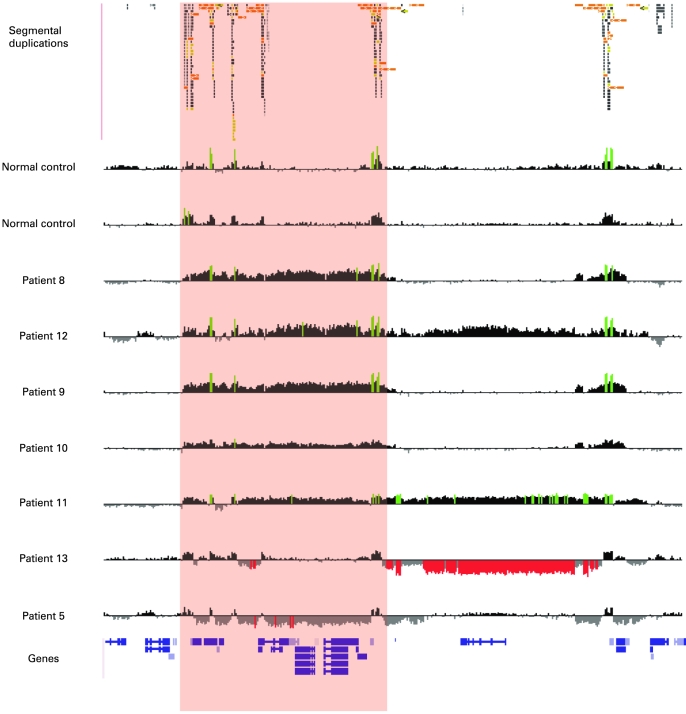
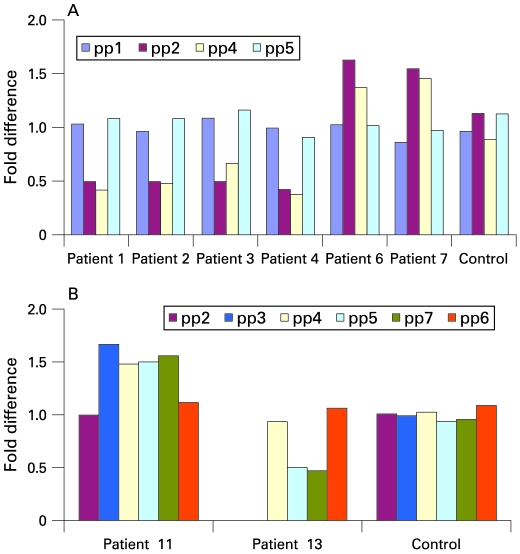
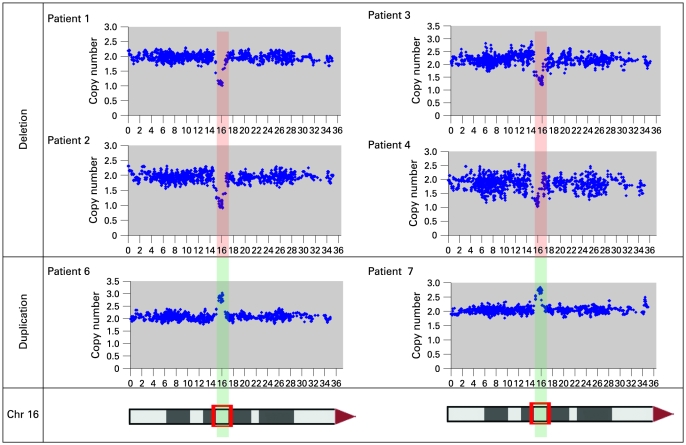
A bacterial artificial chromosome (BAC) array comparative genome hybridisation (CGH) screen of 1027 patients with mental retardation and/or multiple congenital anomalies (MR/MCA) was performed. The BAC array CGH screen identified five patients with deletions and five with apparently reciprocal duplications of 16p13 covering 1.65 Mb, including 15 RefSeq genes. In addition, three atypical rearrangements overlapping or flanking this region were found. Fine mapping by high-resolution oligonucleotide arrays suggests that these deletions and duplications result from non-allelic homologous recombination (NAHR) between distinct LCR16 subunits with >99% sequence identity. Deletions and duplications were either de novo or inherited from unaffected parents. To determine whether these imbalances are associated with the MR/MCA phenotype or whether they might be benign variants, a population of 2014 normal controls was screened. The absence of deletions in the control population showed that 16p13.11 deletions are significantly associated with MR/MCA (p = 0.0048). Despite phenotypic variability, common features were identified: three patients with deletions presented with MR, microcephaly and epilepsy (two of these had also short stature), and two other deletion carriers ascertained prenatally presented with cleft lip and midline defects. In contrast to its previous association with autism, the duplication seems to be a common variant in the population (5/1682, 0.29%).
These findings indicate that deletions inherited from clinically normal parents are likely to be causal for the patients' phenotype whereas the role of duplications (de novo or inherited) in the phenotype remains uncertain. This difference in knowledge regarding the clinical relevance of the deletion and the duplication causes a paradigm shift in (cyto)genetic counselling.
基因组疾病通常由节段性重复序列之间的非等位基因同源重组引起。16号染色体特别富含一种染色体特异性低拷贝重复序列,称为LCR16。
对1027例智力发育迟缓及/或多发先天性畸形(MR/MCA)患者进行了细菌人工染色体(BAC)阵列比较基因组杂交(CGH)筛查。BAC阵列CGH筛查发现5例患者存在16p13缺失,5例存在明显的相互重复,覆盖1.65 Mb,包括15个RefSeq基因。此外,还发现了3个与该区域重叠或侧翼的非典型重排。高分辨率寡核苷酸阵列精细定位表明,这些缺失和重复是由序列同一性>99%的不同LCR16亚基之间的非等位基因同源重组(NAHR)导致的。缺失和重复要么是新发的,要么是从未受影响的父母遗传而来。为了确定这些失衡是否与MR/MCA表型相关,或者它们是否可能是良性变异,对2014名正常对照人群进行了筛查。对照人群中未发现缺失,表明16p13.11缺失与MR/MCA显著相关(p = 0.0048)。尽管表型存在差异,但仍发现了一些共同特征:3例缺失患者表现为智力发育迟缓、小头畸形和癫痫(其中2例还身材矮小),另外2例产前确诊的缺失携带者表现为唇裂和中线缺陷。与之前认为其与自闭症有关的观点不同,该重复似乎是人群中的常见变异(5/1682,0.29%)。
这些发现表明,从临床正常父母遗传而来的缺失可能是患者表型的病因,而重复(新发或遗传)在表型中的作用仍不确定。关于缺失和重复临床相关性的这一知识差异导致了(细胞)遗传咨询的范式转变。