Peters Kate M, Schuman Jason T, Skurray Ronald A, Brown Melissa H, Brennan Richard G, Schumacher Maria A
School of Biological Sciences, A12, University of Sydney, Sydney, NSW, Australia.
Biochemistry. 2008 Aug 5;47(31):8122-9. doi: 10.1021/bi8008246. Epub 2008 Jul 11.
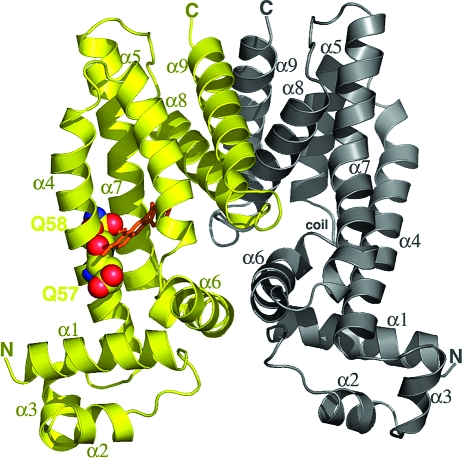
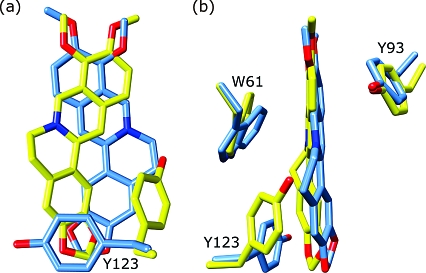
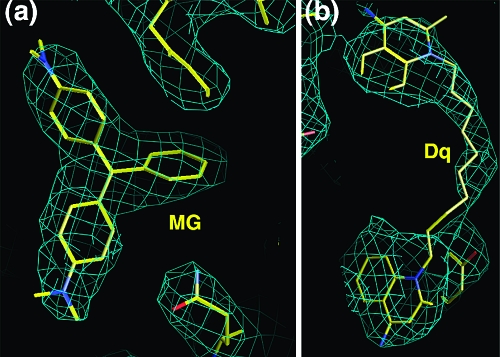
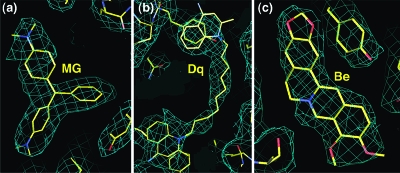
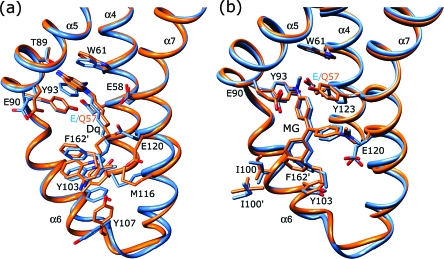
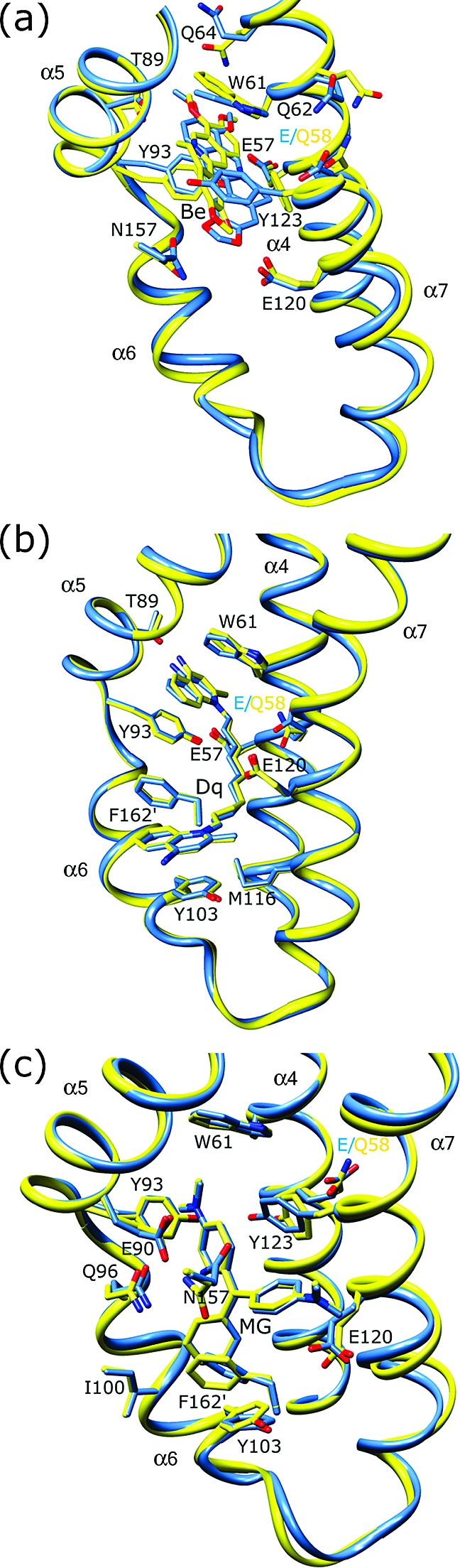
The Staphylococcus aureus multidrug binding protein QacR binds to a broad spectrum of structurally dissimilar cationic, lipophilic drugs. Our previous structural analyses suggested that five QacR glutamic acid residues are critical for charge neutralization and specification of certain drugs. For example, E57 and E58 interact with berberine and with one of the positively charged moieties of the bivalent drug dequalinium. Here we report the structural and biochemical effects of substituting E57 and E58 with alanine and glutamine. Unexpectedly, individual substitutions of these residues did not significantly affect QacR drug binding affinity. Structures of QacR(E57Q) and QacR(E58Q) bound to dequalinium indicated that E57 and E58 are redundant for charge neutralization. The most significant finding was that berberine was reoriented in the QacR multidrug binding pocket so that its positive charge was neutralized by side chain oxygen atoms and aromatic residues. Together, these data emphasize the remarkable versatility of the QacR multidrug binding pocket, illustrating that the capacity of QacR to bind myriad cationic drugs is largely governed by the presence in the pocket of a redundancy of polar, charged, and aromatic residues that are capable of electrostatic neutralization.
金黄色葡萄球菌多药结合蛋白QacR可与多种结构不同的阳离子亲脂性药物结合。我们之前的结构分析表明,五个QacR谷氨酸残基对于电荷中和及某些药物的特异性识别至关重要。例如,E57和E58与小檗碱以及二价药物地喹氯铵的一个带正电荷部分相互作用。在此,我们报告用丙氨酸和谷氨酰胺取代E57和E58的结构和生化效应。出乎意料的是,这些残基的单个取代对QacR药物结合亲和力没有显著影响。与地喹氯铵结合的QacR(E57Q)和QacR(E58Q)的结构表明,E57和E58在电荷中和方面是冗余的。最显著的发现是,小檗碱在QacR多药结合口袋中重新定向,其正电荷被侧链氧原子和芳香族残基中和。总之,这些数据强调了QacR多药结合口袋的显著通用性,表明QacR结合多种阳离子药物的能力在很大程度上取决于口袋中存在能够进行静电中和的极性、带电荷和芳香族残基的冗余结构。