Nagy Alinda, Hegyi Hédi, Farkas Krisztina, Tordai Hedvig, Kozma Evelin, Bányai László, Patthy László
Institute of Enzymology, Biological Research Center, Hungarian Academy of Sciences, H-1113 Budapest, Hungary.
BMC Bioinformatics. 2008 Aug 27;9:353. doi: 10.1186/1471-2105-9-353.
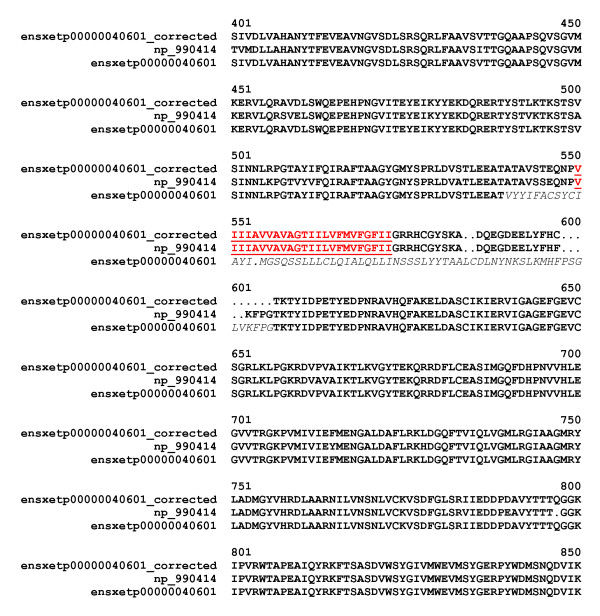
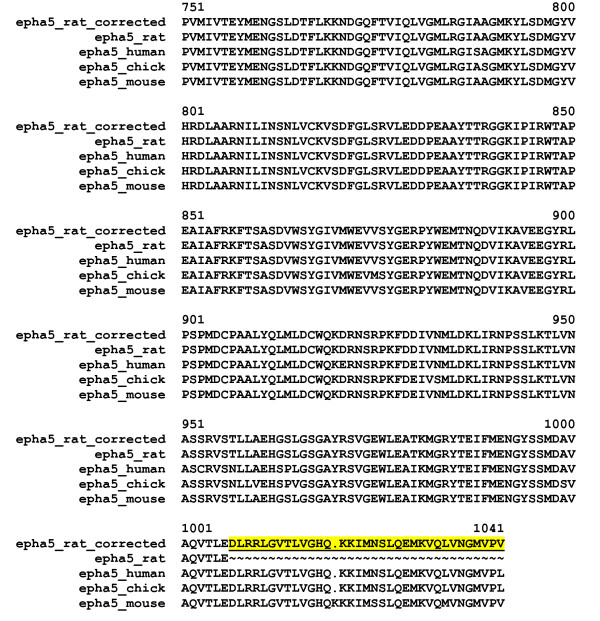
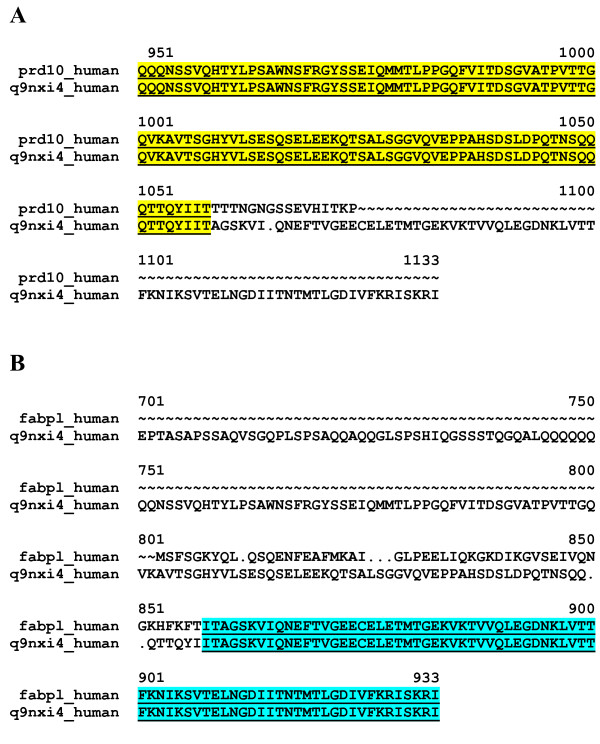
Despite significant improvements in computational annotation of genomes, sequences of abnormal, incomplete or incorrectly predicted genes and proteins remain abundant in public databases. Since the majority of incomplete, abnormal or mispredicted entries are not annotated as such, these errors seriously affect the reliability of these databases. Here we describe the MisPred approach that may provide an efficient means for the quality control of databases. The current version of the MisPred approach uses five distinct routines for identifying abnormal, incomplete or mispredicted entries based on the principle that a sequence is likely to be incorrect if some of its features conflict with our current knowledge about protein-coding genes and proteins: (i) conflict between the predicted subcellular localization of proteins and the absence of the corresponding sequence signals; (ii) presence of extracellular and cytoplasmic domains and the absence of transmembrane segments; (iii) co-occurrence of extracellular and nuclear domains; (iv) violation of domain integrity; (v) chimeras encoded by two or more genes located on different chromosomes.
Analyses of predicted EnsEMBL protein sequences of nine deuterostome (Homo sapiens, Mus musculus, Rattus norvegicus, Monodelphis domestica, Gallus gallus, Xenopus tropicalis, Fugu rubripes, Danio rerio and Ciona intestinalis) and two protostome species (Caenorhabditis elegans and Drosophila melanogaster) have revealed that the absence of expected signal peptides and violation of domain integrity account for the majority of mispredictions. Analyses of sequences predicted by NCBI's GNOMON annotation pipeline show that the rates of mispredictions are comparable to those of EnsEMBL. Interestingly, even the manually curated UniProtKB/Swiss-Prot dataset is contaminated with mispredicted or abnormal proteins, although to a much lesser extent than UniProtKB/TrEMBL or the EnsEMBL or GNOMON-predicted entries.
MisPred works efficiently in identifying errors in predictions generated by the most reliable gene prediction tools such as the EnsEMBL and NCBI's GNOMON pipelines and also guides the correction of errors. We suggest that application of the MisPred approach will significantly improve the quality of gene predictions and the associated databases.
尽管基因组的计算注释有了显著改进,但公共数据库中异常、不完整或预测错误的基因和蛋白质序列仍然大量存在。由于大多数不完整、异常或预测错误的条目并未如此标注,这些错误严重影响了这些数据库的可靠性。在此,我们描述了MisPred方法,它可能为数据库的质量控制提供一种有效手段。MisPred方法的当前版本使用五个不同的程序,基于如果一个序列的某些特征与我们当前关于蛋白质编码基因和蛋白质的知识相冲突,那么该序列可能是错误的这一原则,来识别异常、不完整或预测错误的条目:(i)蛋白质预测的亚细胞定位与相应序列信号缺失之间的冲突;(ii)存在细胞外和细胞质结构域但不存在跨膜区段;(iii)细胞外和核结构域同时出现;(iv)违反结构域完整性;(v)由位于不同染色体上的两个或更多基因编码的嵌合体。
对九种后口动物(智人、小家鼠、褐家鼠、家鼩、原鸡、热带爪蟾、红鳍东方鲀、斑马鱼和玻璃海鞘)和两种原口动物(秀丽隐杆线虫和黑腹果蝇)的预测EnsEMBL蛋白质序列进行分析后发现,预期信号肽的缺失和结构域完整性的违反占大多数预测错误。对NCBI的GNOMON注释管道预测的序列进行分析表明,预测错误率与EnsEMBL的相当。有趣的是,即使是经过人工整理的UniProtKB/Swiss-Prot数据集也被预测错误或异常的蛋白质污染,尽管程度远低于UniProtKB/TrEMBL或EnsEMBL或GNOMON预测的条目。
MisPred在识别由最可靠的基因预测工具(如EnsEMBL和NCBI的GNOMON管道)生成的预测错误方面工作高效,并且还指导错误的纠正。我们建议应用MisPred方法将显著提高基因预测及相关数据库的质量。